
PMMA-g-SOY as a sustainable novel dielectric material†
Abstract
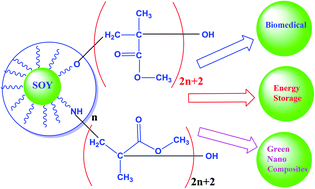
Soy protein (and associated carbohydrate) (SOY) is graft copolymerized with poly(methyl methacrylate) (PMMA) to synthesize novel low cost dielectric materials for multifunctional applications. Graft copolymerization of methyl methacrylate onto pre-activated SOY is carried out using a simple reflux method to form covalently bonded PMMA-g-SOY copolymers. The resulting PMMA-g-SOY is processed into films without employing any toxic chemical solvents. The PMMA-g-SOY films exhibited enhanced storage modulus and a low loss tangent together with promising dielectric properties compared to the pristine PMMA polymer. This strategy may open a new avenue to efficiently use green co-products for multifunctional applications in traditional and structural capacitors.
 Please wait while we load your content...
Please wait while we load your content...

