
High strength biocompatible PEG single-network hydrogels†
Abstract
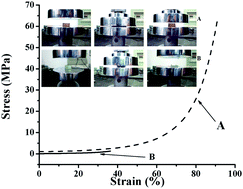
In this work, PEG-based single-chain hydrogels with extremely high strength were successfully prepared via precise design and control over the molecular topology of the polymeric network. Initially, well-defined PEG macromolecules with uniformly dispersed pendant alkynyl groups (PEGn(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH))m on their main chains were synthesized via amine-epoxy chain extension reaction of α,ω-diepoxy PEG and propargylamine. The subsequent copper(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition (CuAAC) of (PEGn(CCH))m and α,ω-diazido PEGn (PEGn(N3)2) gave rise to tough PEG-based hydrogels (Gel-PEGn(N3)2-(PEGn(CCH))m). The lattice size of the Gel-PEGn(N3)2-(PEGn(CCH))m networks can be tailored by varying chain lengths of PEG repeating segments in (PEGn(CCH))m and PEGn(N3)2. Different from traditional PEG hydrogels prepared by CuAAC, such as hydrogels from tetrakis(2-propynyloxymethyl)methane and PEGn(N3)2, the current novel hydrogels possess not only a high mechanical strength up to 62.5 MPa, but are also biodegradable favored by the presence of triethylamine groups in the (PEGn(CCH))m macromolecules. Furthermore, excellent biocompatibility of the Gel-PEGn(N3)2-(PEGn(CCH))m was demonstrated according to the in vitro cytotoxicity assay. Hence, it can be ascertained that Gel-PEGn(N3)2-(PEGn(CCH))m has promising potential for artificial medical devices or scaffolding materials for regenerative medicine.
CH))m on their main chains were synthesized via amine-epoxy chain extension reaction of α,ω-diepoxy PEG and propargylamine. The subsequent copper(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition (CuAAC) of (PEGn(CCH))m and α,ω-diazido PEGn (PEGn(N3)2) gave rise to tough PEG-based hydrogels (Gel-PEGn(N3)2-(PEGn(CCH))m). The lattice size of the Gel-PEGn(N3)2-(PEGn(CCH))m networks can be tailored by varying chain lengths of PEG repeating segments in (PEGn(CCH))m and PEGn(N3)2. Different from traditional PEG hydrogels prepared by CuAAC, such as hydrogels from tetrakis(2-propynyloxymethyl)methane and PEGn(N3)2, the current novel hydrogels possess not only a high mechanical strength up to 62.5 MPa, but are also biodegradable favored by the presence of triethylamine groups in the (PEGn(CCH))m macromolecules. Furthermore, excellent biocompatibility of the Gel-PEGn(N3)2-(PEGn(CCH))m was demonstrated according to the in vitro cytotoxicity assay. Hence, it can be ascertained that Gel-PEGn(N3)2-(PEGn(CCH))m has promising potential for artificial medical devices or scaffolding materials for regenerative medicine.
 Please wait while we load your content...
Please wait while we load your content...

