Intrinsic carrier mobility of germanene is larger than graphene's: first-principle calculations
Xue-Sheng
Ye
a,
Zhi-Gang
Shao
*a,
Hongbo
Zhao
a,
Lei
Yang
ab and
Cang-Long
Wang
a
aLaboratory of Quantum Engineering and Quantum Materials, SPTE, South China Normal University and Institute of Modern Physics, Chinese Academy of Sciences, Guangzhou 510006, China. E-mail: zgshao@scnu.edu.cn
bDepartment of Physics, Lanzhou University, Lanzhou 730000, China
First published on 26th March 2014
Abstract
Shown here is the intrinsic carrier mobility (ICM) of germanene, a group-IV graphene-like two-dimensional buckled nanosheet. Specifically, combining the Boltzmann transport equation with the relaxation time approximation at the first-principle level, it was calculated that the ICM of the germanene sheet can reach ∼6 × 105 cm2 V−1 s−1, in an order of magnitude (105 cm2 V−1 s−1), and even larger than that of graphene. The high ICM of germanene is attributed to the large buckled distance and the small effective mass. Since Ge has good compatibility with Si in the conventional semiconductor industry, the results indicate that germanene should be a good supplement to prospective nanoelectronics.
1 Introduction
Graphene, the two-dimensional (2D) honeycomb network of carbon atoms, has become the most investigated material due to its fascinating properties, such as its linearly dispersing electronic bands at Fermi level contributing to its high carrier mobility.1–3 The group IV elements graphene-like 2D sheets, such as those formed from Si and Ge, named silicene and germanene, respectively, have attractive fundamental physical and chemical properties,4–10 and can easily fit into the silicon-based electronic industry. Therein, silicene has been synthesized by the epitaxial growth of Si on Ag(110), and Ag(111).4,11–17 Recently, germanene has also attracted intense studies, where germanene with a low-buckled structure has been predicted to be stable5,18–24 and semimetallic.7 Lately, Bianco et al. have synthesized and characterized the germanane (hydrogenated germanene).3,21 What is more, since the exciton Bohr radius (23.4 nm) in bulk Ge crystal is much longer than that (4.9 nm) in bulk Si crystal,25,26 Ge nanomaterials will be more affected by those sizes than Si nanomaterials.25 Due to its good compatibility with Si and high carrier mobility, Ge has potential advantages for improving performance of silicon-based electronic devices.4,6–8,27,28As a matter of fact, intrinsic carrier mobility (ICM) of materials is the crucial factor for semiconducting materials.6,29–31 Previously, the first-principle methodology, developed by the group of Shuai, incorporating the density functional theory, the Boltzmann transport equation, and the deformation potential (DP) theory has been carried out to predict ICM of many organic materials.30–35 We have predicted that the ICM of silicene reaches 2.5 × 10 cm2 V−1 s−1 with the same first-principle methodology.36 In addition, theoretical calculations show that germanene also has graphene-like electronic band structure, resulting in charge carriers behaving as massless Dirac fermions. Moreover, the buckled structure leads to reduction of electron–phonon coupling strength. Therefore, the buckled structure should affect the ICM of germanene strongly.
Hence, we predict the ICM of germanene using the first-principle methodology incorporating band structure calculations with the density functional theory and the Boltzmann transport equation under the DP theory.30–35 We have also investigated the influence of buckled structure on the ICM of germanene. Our results can shed new light on ICM of materials and be used as a guide for further experimental nanoelectronics.
2 Model and methods
Fig. 1 shows the low-buckled structure of germanene. In order to present more intuitive explanation for transport property, we build a super-cell along two vertical directions a and b in the charge transport calculation.32a and b are the directions of dilation. Dark dashed lines label the rectangle supercell, where the lattice constants are a0 = 4.03 Å and b0 = 6.97 Å at equilibrium geometry. Geometry optimization and the band structure calculations are performed using density-functional theory as implemented in CASTEP package.37 Perdew–Burke–Ernzerhof (PBE) functional38 within the generalized gradient approximation (GGA) is taken into account to deal with exchange and correlation term. For germanene, a plane wave basis set with the energy cutoff of 500 eV and Vanderbilt ultrasoft pseudopotential39 are applied. The Brillouin zone is sampled by a 49 × 49 × 1 Monkhorst–Pack mesh of k-point.40 The vacuum layer thickness is 16 Å. The calculated bond length and low-buckled distance of germanene are 2.42 Å and 0.69 Å, respectively, which are in good agreement with the calculated values in ref. 5. The low-buckled distance of germanene is higher than that of silicene (0.37–0.46 Å).5,41,42 | ||
| Fig. 1 Side (a) and top (b) views of schematic diagram of the germanene sheet. a and b are the directions of dilation. The rectangle supercell (drawn with dark dashed line) are labeled. | ||
According to the aforementioned first-principle method,30–36 the relaxation time of carriers of germanene can be expressed as
 | (1) |
Incorporating the Boltzmann transport theory with relaxation time approximation, the ICM μ can be derived as
 | (2) |
3 Results and discussion
The band structure and density of states (DOS) of the germanene sheet are shown in Fig. 2. The k-point separation of 10−3 Å−1 on the Brillouin zone path is taken to calculate accurately the group velocity.36,40,43,44 It can be seen from the Fig. 2 that the germanene has Dirac cones, which is consistent with previous studies.5,19,45,46 The linearly dispersing electronic bands at the Fermi level causes the carriers to behave as Dirac fermions17 at the speed about 3.8 × 105 m s−1.47 For the transport calculation, we dilate the super-cell of germanene along the axis a or b in the range of ±1.5%.30–34 In order to obtain the relaxation time of germanene, we have calculated the Fermi level shift ΔE and the total energy (E) of the super-cell as functions of the dilation. The relationship between the Fermi level shift ΔE and the dilation is plotted in Fig. 3(a), which is fitted linearly as ΔE = E1(Δl/l0). And the relationship between total energy and the dilation is plotted in Fig. 3(b), which is fitted parabolically as (E − E0)/S0 = Cα(Δl/l0)2/2. Cα is the elastic constant and Δl/l0 describes the dilation. E0, S0, and l0 are the total energy, cell area, and lattice constant, respectively.30–36 | ||
| Fig. 2 Band structure and DOS of the germanene sheet. | ||
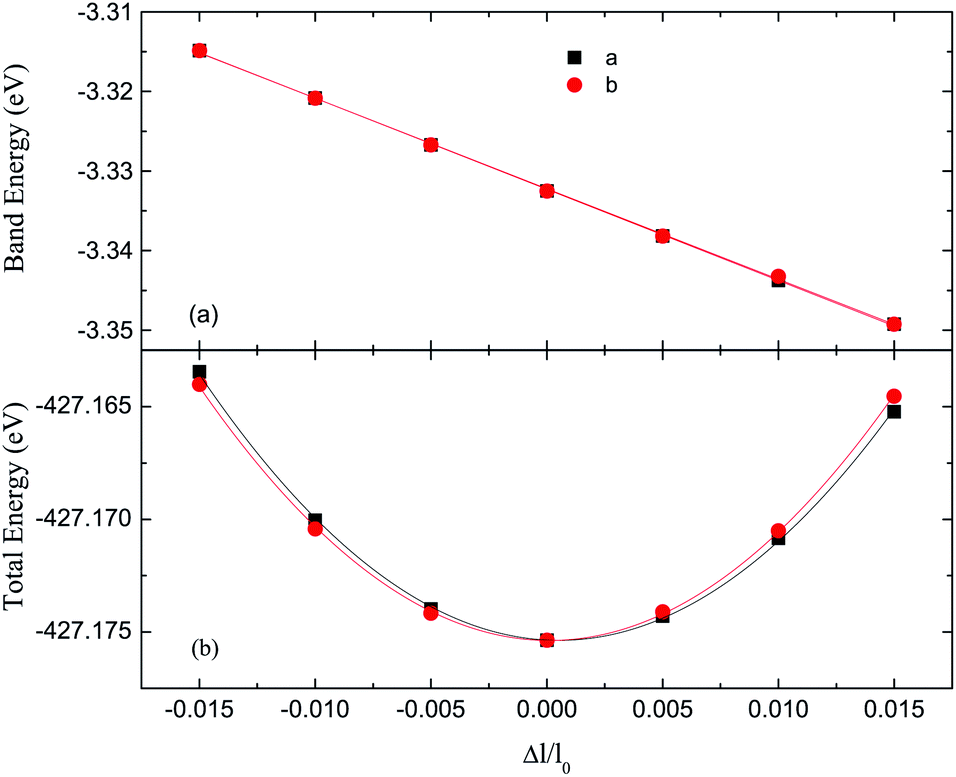 | ||
| Fig. 3 Fermi level shift (a) and the total energy (b) as functions of the lattice dilation Δl/l0 along the directions of a and b for germanene. The linear fit gives the DP constant and the parabola fit gives the elastic constant, respectively. | ||
We obtain the DP constant E1 by linearly fitting the data in Fig. 3(a) and the elastic constant Cα by the parabola fitting the data in Fig. 3(b).30–36 With eqn (1) and (2), we calculate the relaxation time and ICM of germanene. The relevant results are summarized in Table 1. We also provide the relevant results of graphene and silicene calculated in previous studies30–36 for comparison.
| System | BP | Δh | Axis | L | E 1 | C α | τ eD | τ hD | μ e | μ h |
|---|---|---|---|---|---|---|---|---|---|---|
| Germanene | 2.04 | 0.69 | a | 4.03 | 1.16 | 56.01 | 5.26 | 5.46 | 6.09 | 6.39 |
| b | 6.97 | 1.15 | 55.98 | 5.39 | 5.58 | 6.24 | 6.54 | |||
| Silicene36 | 2.73 | 0.44 | a | 3.88 | 2.13 | 86.48 | 1.84 | 1.84 | 2.58 | 2.23 |
| b | 6.71 | 2.13 | 85.99 | 1.83 | 1.83 | 2.57 | 2.22 | |||
| Graphene33 | 3.05 | 0 | a | 2.46 | 5.14 | 328.02 | 2.24 | 2.27 | 3.39 | 3.22 |
| b | 4.26 | 5.00 | 328.30 | 2.37 | 2.40 | 3.20 | 3.51 |
The buckled distances of graphene, silicene, and germanene increase successively.5,41 It can be seen from Table 1 that the elastic constant decreases as the buckled distance increases. The elastic constant of graphene with no buckled structure is the largest compared with the smallest value of germanene with the highest buckled structure. This result is consistent with the mechanical properties of germanene investigated using the Quantum-ESPRESSO package recently, where Si–Si bonds of silicene and Ge–Ge bonds of germanene are more flexible than the C–C ones of graphene with the buckled structure.41 In order to explain how the low buckled distance affects the elastic constant, we calculated bond population of the three honeycomb structures by CASTEP code. It is shown in Table 1 that the larger buckled distance leads to a smaller bond population. As the bond population increases, the bond strength increases.48 As a consequence, the larger buckled distance leads to the smaller elastic constant. Moreover, it also can be found from Table 1 the DP constant decreases as the buckled distances increase. Due to the buckled structure, when the lattice deformation is applied to the silicene and germanene, the bonds are not directly dilated, which can only cause a small band energy shift. In contrast, when the deformation is applied to graphene with no buckled structure, the dilation has a direct influence on the bond length, so the corresponding band energy shift is large. A similar explanation has been applied to analyze the difference between the graphynes and graphene.34
According to eqn (1), the relaxation time is proportional to the elastic constant and inversely proportional to the square of the DP constant. Substituting the data in Table l, the values of 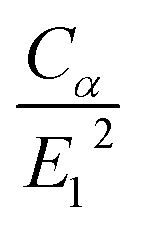 are 12.42, 19.06, and 41.74 for graphene, silicene, and germanene, respectively. As a consequence, the relaxation time around the Dirac cone of germanene is larger than those of silicene and graphene. The relaxation time of graphene is larger than that of silicene, which is just because the shape of the Dirac cone of graphene is more symmetric than that of silicene. It can be seen from eqn (2) the ICM depends not only on the relaxation time but also on group velocities and shapes of the Fermi surfaces. However, the group-IV elements graphene-like 2D sheets all have Dirac cones in their Brillouin zones and they all have high Fermi velocities around the Dirac cone about 6.3 × 105 m s−1, 5.1 × 105 m s−1, and 3.8 × 105 m s−1 for graphene, silicene, and germanene, respectively.47 Therefore, the large relaxation time around the Dirac cone should lead to the high ICM of germanene, shown as in Table 1. In addition, Bardeen and Shockley have pointed out that higher mobilities of electrons and holes in Ge as compared with Si are correlated with a smaller shift of energy gap with dilation,49 which verifies the high ICM of germanene in our results from another perspective.
are 12.42, 19.06, and 41.74 for graphene, silicene, and germanene, respectively. As a consequence, the relaxation time around the Dirac cone of germanene is larger than those of silicene and graphene. The relaxation time of graphene is larger than that of silicene, which is just because the shape of the Dirac cone of graphene is more symmetric than that of silicene. It can be seen from eqn (2) the ICM depends not only on the relaxation time but also on group velocities and shapes of the Fermi surfaces. However, the group-IV elements graphene-like 2D sheets all have Dirac cones in their Brillouin zones and they all have high Fermi velocities around the Dirac cone about 6.3 × 105 m s−1, 5.1 × 105 m s−1, and 3.8 × 105 m s−1 for graphene, silicene, and germanene, respectively.47 Therefore, the large relaxation time around the Dirac cone should lead to the high ICM of germanene, shown as in Table 1. In addition, Bardeen and Shockley have pointed out that higher mobilities of electrons and holes in Ge as compared with Si are correlated with a smaller shift of energy gap with dilation,49 which verifies the high ICM of germanene in our results from another perspective.
Moreover, the ICM can be expressed as 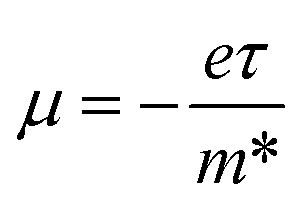 . As the mean scattering time τ increases or the effective mass m* decreases, the ICM increases.33 In ref. 6, Ni et al. have shown the effective mass of germanene is mKΓe = 0.014 m0 and mMKe = 0.029 m0 at E⊥ = 0.4 V Å−1 is close to the value of bilayer graphene and smaller then the experimental value me = 0.06 m0 of graphene by Novoselov et al.1 Hence it is reasonable that the calculated ICM of germanene is larger than that of graphene.
. As the mean scattering time τ increases or the effective mass m* decreases, the ICM increases.33 In ref. 6, Ni et al. have shown the effective mass of germanene is mKΓe = 0.014 m0 and mMKe = 0.029 m0 at E⊥ = 0.4 V Å−1 is close to the value of bilayer graphene and smaller then the experimental value me = 0.06 m0 of graphene by Novoselov et al.1 Hence it is reasonable that the calculated ICM of germanene is larger than that of graphene.
4 Conclusions
In summary, we have predicted the intrinsic carrier mobility (ICM) of germanene based on band structure calculations with the density functional theory, and Boltzmann transport equation coupled with the deformation potential theory at the first-principle level. The ICM of germanene can reach 6.24 × 105 cm2 V−1 s−1 and 6.54 × 105 cm2 V−1 s−1 for electrons and holes, respectively, at room temperature. Our results show that calculated ICM of germanene is in the same order of magnitude of that in graphene and silicene, which originates from the effect of the buckled distance and the small effective mass. From Table 1, it is clear that the ICM of germanene is even larger than that of graphene and silicene. Within the DP theory, we only take the scattering of a thermal electron or hole by acoustic phonon into account.33 Whatever the circumstances, it is remarkable that the ICM of germanene is in the order of magnitude of 105 cm2 V−1 s−1. Recently, some studies by first-principle calculations have demonstrated that graphene and h-BN are suitable substrates to synthesize germanene.23,24 For example, Cai et al. show germanene can be stable and preserve its linear energy and low-buckled structure on the substrate of graphene.24 And, Li et al. show that germanene can stably attach on h-BN substrate via van der Waals interactions and the high carrier mobility of germanene can be well preserved in that situation.23 The most important thing is that the substrate can open the bandgap in germanene.6,23,24 So germanene is suitable for the semiconductor industry. Nevertheless, for real applications in nanoelectronics the ICM of germanene with the induced substrate needs further research. In any event, since it has a high ICM and good compatibility with Si, germanene should be a good supplement in nanoelectronics.Acknowledgements
Zhi-Gang Shao acknowledges the support by the National Natural Science Foundation of China (Grant no. 11105054 and 11274124) and PCSIRT (Grant no. IRT1243) and by the high-performance computing platform of South China Normal University. Hongbo Zhao acknowledges the support by the National Natural Science Foundation of China (Grant no. 91026005). Lei Yang acknowledged also the support by the National Natural Science Foundation of China (Grant no. 11074077) and the “Strategic Priority Research Program” of the Chinese Academy of Sciences (Grant no. XDA03030100). Cang-Long Wang acknowledges the support by the National Natural Science Foundation of China (Grant no. 11304324).References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Frisov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Frisov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed
- E. Bianco, S. Butler, S. Jiang, O. D. Restrepo, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 4414–4421 CrossRef CAS PubMed
- P. De Padova, C. Quaresima, C. Ottaviani, P. M. Sheverdyaeva, P. Moras, C. Carbone, D. Topwal, B. Olivieri, A. Kara, H. Oughaddou, B. Aufray and G. Le Lay, Appl. Phys. Lett., 2010, 96, 261905 CrossRef PubMed
- S. Cahangirov, M. Topsakal, E. Aktürk, H. Şahin and S. Ciraci, Phys. Rev. Lett., 2009, 102, 236804 CrossRef CAS
- Z. Ni, Q. Liu, K. Tang, J. Zheng, J. Zhou, R. Qin, Z. Gao, D. Yu and J. Lu, Nano Lett., 2012, 12, 113–118 CrossRef CAS PubMed
- M. Houssa, G. Pourtois, V. Afanasev and A. Stesmans, Appl. Phys. Lett., 2010, 96, 082111 CrossRef PubMed
- X.-Q. Wang, H.-D. Li and J.-T. Wang, Phys. Chem. Chem. Phys., 2012, 14, 3031–3036 RSC
- L. Stille, C. J. Tabert and E. J. Nicol, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 195405 CrossRef
- L. Matthes, O. Pulci and F. Bechstedt, J. Phys.: Condens. Matter, 2013, 25, 395305 CrossRef PubMed
- B. Aufray, A. Kara, S. Vizzini, H. Oughaddou, C. Leandri, B. Ealet and G. Le Lay, Appl. Phys. Lett., 2010, 96, 183102 CrossRef PubMed
- B. Lalmi, H. Oughaddou, H. Enriquez, A. Kara, S. Vizzini, B. Ealet and B. Aufray, Appl. Phys. Lett., 2010, 97, 223109 CrossRef PubMed
- A. Kara, H. Enriquez, A. P. Seitsonen, L. Lew Yan Voon, S. Vizzini, B. Aufray and H. Oughaddou, Surf. Sci. Rep., 2012, 67, 1–18 CrossRef CAS PubMed
- P. De Padova, O. Kubo, B. Olivieri, C. Quaresima, T. Nakayama, M. Aono and G. Le Lay, Nano Lett., 2012, 12, 5500–5503 CrossRef CAS PubMed
- B. Feng, Z. Ding, S. Meng, Y. Yao, X. He, P. Cheng, L. Chen and K. Wu, Nano Lett., 2012, 12, 3507–3511 CrossRef CAS PubMed
- P. Vogt, P. De Padova, C. Quaresima, J. Avila, E. Frantzeskakis, M. C. Asensio, A. Resta, B. Ealet and G. Le Lay, Phys. Rev. Lett., 2012, 108, 155501 CrossRef
- L. Chen, C.-C. Liu, B. Feng, X. He, P. Cheng, Z. Ding, S. Meng, Y. Yao and K. Wu, Phys. Rev. Lett., 2012, 109, 056804 CrossRef
- G. G. Guzmán-Verri and L. C. Lew Yan Voon, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 075131 CrossRef
- C.-C. Liu, W. Feng and Y. Yao, Phys. Rev. Lett., 2011, 107, 076802 CrossRef
- F. Bechstedt, L. Matthes, P. Gori and O. Pulci, Appl. Phys. Lett., 2012, 100, 261906 CrossRef PubMed
- K. J. Koski and Y. Cui, ACS Nano, 2013, 7, 3739–3743 CrossRef CAS PubMed
- D. Kaltsas and L. Tsetseris, Phys. Chem. Chem. Phys., 2013, 15, 9710–9715 RSC
- L. Li and M. Zhao, Phys. Chem. Chem. Phys., 2013, 15, 16853–16863 RSC
- Y. Cai, C.-P. Chuu, C. M. Wei and M. Y. Chou, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 245408 CrossRef
- H. Zhou, M. Zhao, X. Zhang, W. Dong, X. Wang, H. Bu and A. Wang, J. Phys.: Condens. Matter, 2013, 25, 395501 CrossRef PubMed
- Y. Wu and P. Yang, Chem. Mater., 2000, 12, 605–607 CrossRef CAS
- Q. Pang, Y. Zhang, J.-M. Zhang, V. Ji and K.-W. Xu, Nanoscale, 2011, 3, 4330–4338 RSC
- W. Wei, Y. Dai, B. Huang and T. Jacob, Phys. Chem. Chem. Phys., 2013, 15, 8789–8794 RSC
- J. Wang, R. Zhao, M. Yang, Z. Liu and Z. Liu, J. Chem. Phys., 2013, 138, 084701 CrossRef PubMed
- M.-Q. Long, L. Tang, D. Wang, L. Wang and Z. Shuai, J. Am. Chem. Soc., 2009, 131, 17728–17729 CrossRef CAS PubMed
- Z. Shuai, L. Wang and Q. Li, Adv. Mater., 2011, 23, 1145–1153 CrossRef CAS PubMed
- M. Long, L. Tang, D. Wang, Y. Li and Z. Shuai, ACS Nano, 2011, 5, 2593–2600 CrossRef CAS PubMed
- J. Xi, M. Long, L. Tang, D. Wang and Z. Shuai, Nanoscale, 2012, 4, 4348–4369 RSC
- J. Chen, J. Xi, D. Wang and Z. Shuai, J. Phys. Chem. Lett., 2013, 4, 1443–1448 CrossRef CAS
- J. Chen, D. Wang and Z. Shuai, J. Chem. Theory Comput., 2012, 8, 3338–3347 CrossRef CAS
- Z.-G. Shao, X.-S. Ye, L. Yang and C.-L. Wang, J. Appl. Phys., 2013, 114, 093712 CrossRef PubMed
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS
- D. Vanderbilt, Phys. Rev. B: Condens. Matter, 1990, 41, 7892–7895 CrossRef
- J. D. Pack and H. J. Monkhorst, Phys. Rev. B: Solid State, 1977, 16, 1748–1749 CrossRef
- T. Kaloni and U. Schwingenschlögl, Chem. Phys. Lett., 2013, 583, 137–140 CrossRef CAS PubMed
- F.-B. Zheng and C.-W. Zhang, Nanoscale Res. Lett., 2012, 7, 1–5 CrossRef PubMed
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169–11186 CrossRef CAS
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- K. Takeda and K. Shiraishi, Phys. Rev. B: Condens. Matter, 1994, 50, 14916–14922 CrossRef CAS
- H. Behera and G. Mukhopadhyay, AIP Conf. Proc., 2011, 1349, 823–824 CrossRef CAS PubMed
- L. C. Lew Yan Voon, E. Sandberg, R. S. Aga and A. A. Farajian, Appl. Phys. Lett., 2010, 97, 163114 CrossRef PubMed
- M. D. Segall, R. Shah, C. J. Pickard and M. C. Payne, Phys. Rev. B: Condens. Matter, 1996, 54, 16317–16320 CrossRef CAS
- J. Bardeen and W. Shockley, Phys. Rev., 1950, 80, 72–80 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2014 |