A smart synthesis of gold/polystyrene core–shell nanohybrids using TEMPO coated nanoparticles†
Katarzyna Zawadaa,
Waldemar Tomaszewskib and
Elżbieta Megiel*c
aMedical University of Warsaw, Faculty of Pharmacy with the Laboratory Medicine Division, Banacha 1, 02-097, Warsaw, Poland
bWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, 00-664 Warsaw, Poland
cUniversity of Warsaw, Faculty of Chemistry, Pasteura 1, 02-093 Warsaw, Poland. E-mail: emegiel@chem.uw.edu.pl
First published on 23rd May 2014
Abstract
This paper describes a novel route to the synthesis of core–shell nanostructures utilizing Nitroxide Mediated Radical Polymerization (NMRP). The hybrid nanostructures with nanogold cores and precisely designed polystyrene shells (PDI < 1.3) have been synthesized through late injection of TEMPO coated gold nanoparticles into a TEMPOL (4-hydroxy-TEMPO) mediated styrene polymerization system. Thermal analyses have shown that the thus obtained polystyrene modified gold nanoparticles are significantly more stable to the thermal treatment than only thiol-stabilized gold nanoparticles (aggregation has not been observed even at 300 °C). The obtained materials exhibit intensive surface plasmon resonance (SPR) observed near 520 nm that provides an opportunity to apply them as high-temperature optical sensors. The proposed procedure allows for precise synthesis of tailor-made nanohybrids via macroradical coupling.
1. Introduction
Over the past decade, core–shell nanostructures with metal nanoparticle cores and polymer shells have attracted great attention as novel materials that combine the unique properties of nanoparticles with useful characteristics of polymers.1,2 Gold nanoparticles (AuNPs) are the most stable metal nanoparticles and they are extensively studied due to their fascinating quantum-size-related properties as well as because of unique biological and catalytic activity.3 One of the most important size-related phenomena observed for AuNPs is the surface plasmon resonance (SPR)4–6 resulting from the strong interactions of free conduction electrons with visible light manifesting as the collective oscillation with respect to the nanoparticle lattice in resonance with the light field. The SPR frequency depends strongly on the size and shape of nanoparticles,7 dielectric properties of the surrounding chemical environment7 and inter-nanoparticle coupling interactions.8,9 This phenomenon is leading AuNPs to potential applications in highly sensitive chemical and biological sensors.3,10 However, it is a well-known problem that the metal nanoparticles show high surface energy which results in agglomeration tendency.2 The excellent chemical stability of gold nanoparticles is crucial for their wide applications in novel materials designing. Formation of polymer brushes via tethering polymer chains onto the nanoparticles surface allows for improvement in nanoparticles stability and, at the same time, leads to obtaining a new type of material combining the different characteristics of the components.11 In general, the same types of approach are used for tethering polymer chains onto nanoparticles, nanotubes or flat surfaces.2 Currently, there are two main methods towards tethering of polymer chains onto a solid surface: reversible physisorption and irreversible covalent attaching by chemical bonding.12 Nevertheless, reversible physisorption results in obtaining less stable polymer brushes due to existence of only weak interactions between polymer and the surface.13 Stable tethered polymer shells may be obtained using covalent attaching including “grafting to” and “grafting from” methods.14 The “grafting to” approach is based on the reaction with reactive groups localized on the surface, to which polymer chains, usually end-functionalized and prepared in advance, are tethered.15 However, “grafting to” approach is limited to preparation of relatively low density polymer shells.16 The high density polymer shells can be formed via “grafting from” approach12 consisting of the surface initiated polymerization wherein the polymerization initiator is immobilized on the surface of the substrate and the polymer chains grow in situ from the flat surface or the surface of a nanoparticle. The living/controlled free radical polymerization (CLRP)17a–f which enables the designing of polymers with narrow molecular weight distribution and complex architecture has been most extensively used in core–shell nanostructures preparation. The predominance of living/controlled free radical polymerization in the core–shell nanostructures preparation over living ionic and ring opening polymerizations results mainly from the lower sensitivity to moisture and impurities,18 compatibility with both aqueous and organic media, and tolerance toward a wide range of functional groups.19 The reversible-addition-fragmentation-chain transfer (RAFT)20 technique is one of the methods of CLRP which is widely used for the preparation of well-defined end-functionalized polymers which can be easily attached to AuNPs surface through the “grafting to” approach.21Functionalized AuNPs have been widely employed in the synthesis of core–shell nanohybrids via surface-initiated controlled polymerization (SI-CRP).19 The first applied method, for this purpose, was atom transfer radical polymerization (ATRP).22 Nuß et al.23 have shown that AuNPs functionalized by an α-bromoester group can be grafted with poly n-butylacrylate by ATRP mechanism. Synthesis of AuNPs coated with well-defined high density poly(methyl methacrylate) shell via surface initiated ATRP was described by Fukuda with co-workers,24 Mandal et al.,25 and at elevated temperature with SiO2 protective shell by Kotal et al.26 The preparation of thermoresponsive, cross-linked poly(N-isopropylacrylamide) coated gold nanoparticles by ATRP mechanism have been described by Kim et al.27
Nevertheless, it has been proven that the ATRP is a suitable technique for preparation of gold nanoparticles coated with polymers. The possibility of the presence of residual amounts of a metal catalyst in the obtained material, which is significant for (bio)medical applications, is worth mentioning.19
Raula et al.28 have reported successful synthesis of gold nanoparticles coated with poly(N-isopropylacrylamide) via surface initiated RAFT polymerization. However, the possible cleavage of RAFT agents from AuNPs surfaces and the tolerance of the RAFT agents to the alkaline or acidic media in this process should be taken into account.9
Nitroxide mediated radical polymerization (NMRP) is based on reversible termination occurring between a growing propagating polymer chain and nitroxide radicals. This type of controlled polymerization is purely thermal process and a catalyst is not required thus additional purification steps for catalyst removal are not needed.29a–c
The grafting of polymer chains via NRMP can be achieved using substrates which surfaces had been modified by conventional radical initiator or nitroxide derivatives in the presence of “free” nitroxide in polymerization system.29d Surface initiated NMRP of styrene has been successfully achieved from TEMPO (2,2,6,6-tetramethypiperidine-1-oxyl) modified: silicon wafers,30a,b silicon oxide surface,30c,d magnetite30e,f or titanium30g nanoparticles, steel flat surface,30h Merrifield resins,30i carbon MWNTs,30j and quantum dots.30k
To the best of our knowledge, grafting of polystyrene chains onto gold nanoparticles surface via NMRP has not been reported yet. The synthesis of TEMPO coated gold nanoparticles and their tethering to gold surface has been described in our previous paper.31
In present work we report a facile synthetic method of preparation of core–shell nanostructures using easy to obtain TEMPO coated gold nanoparticles as precursor. The proposed method is based on a late injection of nitroxide covered gold nanoparticles to 4-hydroxy-TEMPO mediated polymerization of styrene and its further polymerization leading to polymer chains elongation.
The procedure described in this paper can be treated as a novel modification of “grafting to” method allowing for precise designing of grafting density and length of polymer chains tethered to gold surface.
2. Experimental
2.1. Materials
NaBH4, benzoyl peroxide (BPO), 4-hydroxy-TEMPO (TEMPOL), gold(III) chloride trihydrate and all solvents were purchased from Sigma-Aldrich and used as received, styrene (St, Sigma-Aldrich ≥99%) was passed through a basic activated aluminum oxide column before use (Merck, 0.06 mm). Bis(N-oxy-2,2,6,6-tetramethylpiperidyl)-4,5-dithiooctanoate (DiSS) was synthesized according to the procedure described in the literature.322.2. Techniques
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 (w/w), FT-IR spectra of PS-AuNPs were recorded from KBr pellets immersed earlier in THF solution of PS-AuNPs and dried in argon atmosphere.
:1000 (w/w), FT-IR spectra of PS-AuNPs were recorded from KBr pellets immersed earlier in THF solution of PS-AuNPs and dried in argon atmosphere.2.3. Preparation of TEMPO coated gold nanoparticles (T-AuNPs)
In the common procedure, 115 mg of HAuCl4·3H2O (300 μmol) was dissolved in 80 ml ethyl acetate (p.a.), 250 mg of DiSS (579 μmol) was added (Au:S = 1:3.2) and the mixture was stirred for 30 minutes, after which, 30 minute-aged solution of NaBH4 in methanol (200 mg in 50 ml) was slowly added during half hour, under vigorous stirring.
After the mixture turned from light orange through light brown to burgundy red, the solution was stirred for 24 h at room temperature. Following this, 50 mg of HAuCl4·3H2O (130 μmol) dissolved in 10 ml ethyl acetate was drop-wise added to the mixture and mixture was left for the next 24 hours under vigorous stirring. After this time, the mixture was washed with deionized water (3 × 100 ml), the organic phase was evaporated to ca. 5 ml and precipitated by the addition of n-hexane (150 ml). The freshly obtained black precipitate was sonicated in hexane for 1 minute and centrifuged at 15000 rpm for 5 minutes. The solid product was dissolved in THF and then precipitated with hexane. The procedure of dissolving T-AuNPs in THF and precipitating with hexane was repeated three times until no traces of free ligands were found. The absence of impurities was confirmed by TLC. The obtained solid was dried under vacuum at 40 °C. Finally 62 mg of black solid, yield 73% (calculated only for metal core) was obtained.
2.4. Preparation of polystyrene coated gold nanoparticles (AuPS)
TEMPOL (73.3 mg, 0.43 mmol), BPO (34.3 mg, 0.14 mmol) and styrene St (8 ml, 70 mmol) were added to the Schlenk flask sealed with a silicone rubber septum. The flask was degassed by three freeze–pump–thaw cycles and dry argon was introduced to the flask. The ratio of reagents was St:BPO:TEMPOL 500:1:3. The mixture was heated at 120 °C in thermostatically controlled oil bath with constant stirring, using magnetic stirrer, for 24 hours. After 24 hours of polymerization, 1 ml of sample was taken using syringe and then immediately 2 ml of acetone solution containing 20 mg of T-AuNPs (earlier degassed by dry argon bubbling) was transferred into polymerization system in counter flow of argon. After addition of solution of T-AuNPs the mixture turned from colourless to cherry red. The polymerization was continued for 6 h. Two 2 ml samples were collected from the reaction mixture, after 2 and 4 hours respectively, using syringe (in counter flow of argon). During the polymerization time the colour of the mixture did not change which indicated that aggregation processes had not occurred. As a result of polymerization viscous, cherry-red and homogeneous mixture was obtained. In order to isolate grafted polymers (AuPS) from free polymers in solution (PSF) all the obtained samples (collected 2, 4 and 6 hours after T-AuNPs injection) were dissolved in 10 ml of THF and precipitated by the addition of n-hexane (50 ml). The obtained suspensions were centrifuged (15000 rpm, 10 minutes). The dark red solid was dissolved in THF and then precipitated with hexane. The procedure of dissolving in THF and precipitating with hexane was repeated until the addition of excess of methanol to the supernatant did not result in its turbidity. Finally dark red solid products (AuPS2, AuPS4, AuPS6) were dried under vacuum at 40 °C.
2.5. Cleaving of polystyrene chains from polystyrene coated nanoparticles (AuPS) with iodine24
4 mg of the AuPS was dissolved in a 5 mM of iodine solution in dichloromethane (3 ml) and stirred for 17 hours. After this time mixture turned from homogeneous purple to turbid brownish. The mixture was evaporated, dissolved in THF, and centrifuged (15000 rpm, 10 min) to remove the insoluble component. Polystyrene from supernatant was precipitated by the addition of methanol. The obtained suspension was centrifuged and the obtained white solid was washed with methanol, dissolved in THF and then subjected to SEC analysis. The obtained polymers have been denoted as CPS2, CPS4, CPS6.
3. Results and discussion
AuPS nanostructures with nanogold core and polystyrene shell have been synthesized via late injection of TEMPO coated gold nanoparticles into TEMPOL mediated polystyrene polymerization (24 h after beginning). The polymerization continued for three periods of time: 2 h (AuPS2), 4 h (AuPS4) and 6 h (AuPS6) according to Scheme 1. The ratio of reagents was St:BPO:TEMPOL 500:1:3, temperature 120 °C. (according to Scheme 1). T-AuNPs were obtained in one-phase procedure using bisnitroxide disulfide as stabilizing ligand, ethyl acetate as reaction medium and methanol as solvent for NaBH4 (according to Scheme 2).
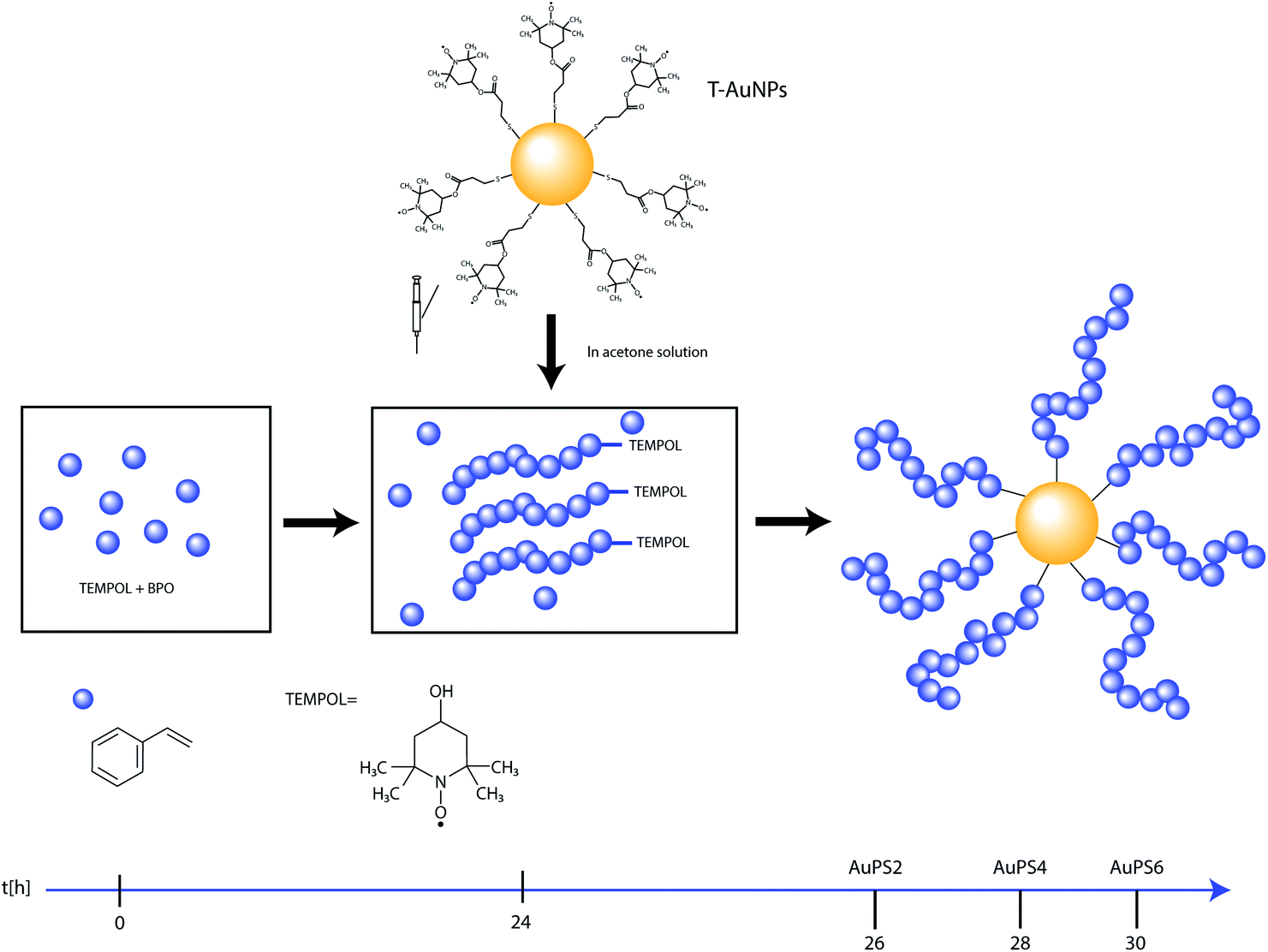 | ||
| Scheme 1 Synthetic route to preparation of hybrids with nanogold core and polystyrene shell (AuPS). | ||
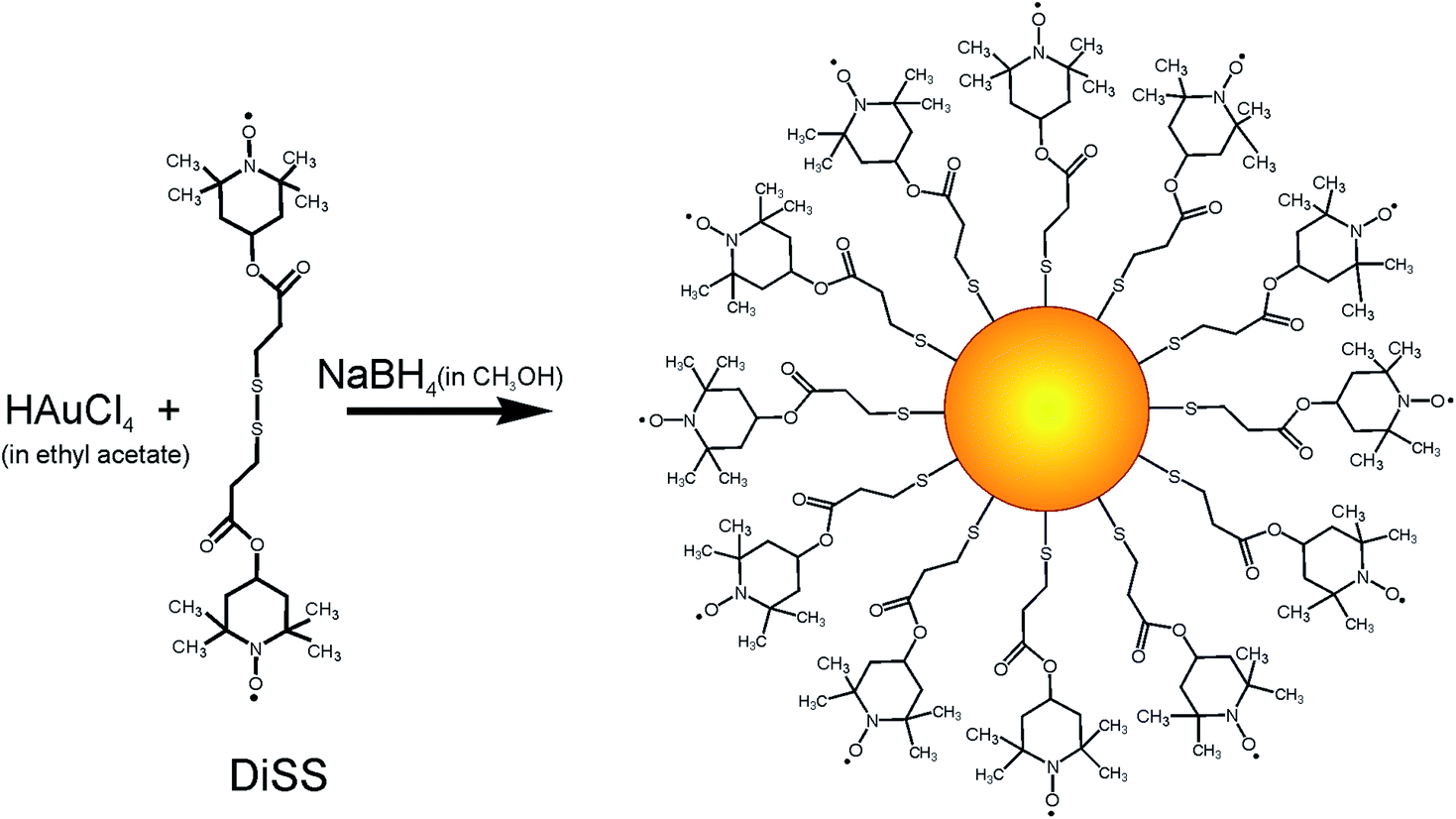 | ||
| Scheme 2 Synthetic route to the T-AuNPs preparation. | ||
The protocol of TEMPO coated gold nanoparticles preparation, according to Scheme 2, had been developed in our laboratory earlier.31 However, we modified this protocol to obtain nanoparticles with larger size, exhibiting stronger Surface Plasmon Resonance (SPR) and simultaneously exhibiting high spin activity. We observed that the time which passed from preparation of methanol solution of NaBH4 to its addition is crucial for reducing power and results in nanoparticles size distribution. Application of 30 minute-aged methanol solution of NaBH4 and additional portion of HAuCl4 solution after 24 hours allows for obtaining nanoparticles with bimodal and narrow size distribution. Apparently, the addition of the next portion of gold salt caused that the synthesis proceeded not only through regular growth but also through nucleation process occurring simultaneously, which lead to creation of small particles population. TEM measurements proved that the obtained material consists of two populations of nanoparticles – larger particles with size 5.69 ± 0.43 nm in diameter, and very small nanoparticles with size 1.56 ± 0.46 nm (Fig. 1).
 | ||
| Fig. 1 TEM image of the obtained T-AuNPs and their size distribution. | ||
Furthermore, Fig. 1 shows the ability of the obtained spin coated gold nanoparticles for self-organization. This phenomenon is clearly visible only for the population of larger nanoparticles. One of the possible explanations of self-assembling ability of the obtained nanoparticles might be the attraction between nanoparticles through interactions between unpaired electrons of paramagnetic molecules and the conduction electrons of gold. It was described earlier that the interactions between the unpaired electrons of nitroxides and the conduction electrons of metal surface leads to the decrease of lifetime of unpaired electrons and as the consequence broadening/suppression of ESR signal of nitroxides adsorbed on the metal surface.33–35 However we haven't observed such an effect in our study. The ESR spectrum of T-AuNPs in acetone (3 mg ml−1) is shown in Fig. 2.
 | ||
| Fig. 2 ESR spectrum of T-AuNPs in acetone at 293 K and simulated spectrum (broad line, triplet component and a sum of both components). | ||
The broad component, dominating the spectrum (95% of total intensity), is the result of averaging of the signals from spin–spin interacting radicals with various coupling constants J in the systems with high radical concentration. Similar domination of the broad single signal has been observed by Chechik et al.36 for thiol-modified gold nanoparticles with high concentration of nitroxides attached to the gold surface. Therefore, the strong prevalence of this component indicates relatively high surface coverage. However, in the case of T-AuNPs obtained in this work, the line is narrower (peak-to-peak line width is 13.0 G in our system vs. 16.8 G for 40 and 14.9 G for 90 nitroxides per nanoparticle reported by Chechik et al.36), suggesting stronger interactions, probably due to higher local concentration. These interactions can be the reason of self-organizing of T-AuNPs as observed by TEM. A triplet with an isotropic 14N-hyperfine splitting of 1.54 mT is typical of a nitroxide radical in solution in a non-polar local environment37,38 and is most probably due to isolated adsorbed radicals. This signal is responsible for 5% of the total intensity. Similar signal has been observed, as dominating, for either low coverage thiol-modified nanoparticles,36 or in our earlier work, for small nanoparticles (average diameter 2.25 nm).31 It can be therefore suggested that this signal is due mainly to radicals adsorbed on small particles, with relatively low nitroxide-nanoparticle ratio, while the broad signal arises from radicals adsorbed in ensembles and thus in higher concentration on large nanoparticles. The reason for such a different ratio can be both the available surface and the surface curvature. Though the concentration of small and large nanoparticles is similar, the amount of radicals adsorbed in both cases would be very different as a result of various types of surface coverage, explaining the small contribution of triplet component to the total ESR intensity.
The broadening of triplet lines (peak-to-peak linewidth 4 G) can be tentatively explained by interactions with a surface of the nanoparticle that this radical is adsorbed on. This effect, already mentioned, is known for small nitroxide radicals adsorbed on the metal surface.38 Another, possible explanation is the interaction between radicals adsorbed on the same nanoparticle, but in much smaller concentration than in case of radicals giving rise to the broad single signal. The broadening of triplet line is rather not the effect of immobilization of the radicals, since this spectrum is symmetrical, and the hindered-tumbling broadening mechanism should affect the linewidth and signal amplitude of the various components asymmetrically.39,40
It is well known, that disulfides, thiols, dithiols and sulfides yield self-assembled monolayers on the surface of many metals (Au, Ag, Cu, Pd, Pt, Ni, Fe).41 Additionally, alkanethiol-coated AuNPs may spontaneously assemble into organized 2D arrays on solid substrates upon solvent evaporation.42 Theoretical and experimental studies have shown that the size and size distribution of nanoparticles, and chain length of the alkylthiol protecting ligand influence this phenomenon.43,44 In our studies a stabilizing ligand can be treated as TEMPO functionalized alkylthiol with short length, hence the prepared nanoparticles should exhibit tendency to self-assembly under evaporation on microscopic grid. The chainlike assembly of larger nanoparticles is clearly visible in Fig. 1. In the case of the smaller nanoparticles significantly lower surface coverage in comparison with coverage of larger ones (see ESR studies) results in weaker intermolecular forces and self-assembly is not clearly observed.
Fig. 3 shows ESR spectra recorded for T-AuNPs and core–shell nanostructures obtained according to Scheme 1. As can be seen, ESR signal completely disappears for AuPS nanostructures. This suggests that polymer chains have been covalently attached to the radicals adsorbed on the gold surface.
 | ||
| Fig. 3 ESR spectra of T-AuNPs (in acetone solution) and polystyrene coated nanoparticles (in THF solution). | ||
Fig. 4 shows UV-Vis spectra (solutions in THF) of T-AuNPs and nanohybrids obtained as a result of polymerization of styrene with T-AuNPs. It is clearly visible that the maximum of plasmon resonance band occurs at the same wavelength of 520 nm. It demonstrates that aggregation of nanoparticles has not occurred and metallic cores of nanohybrids sizes do not increase during the polymerization (carried out at 120 °C).
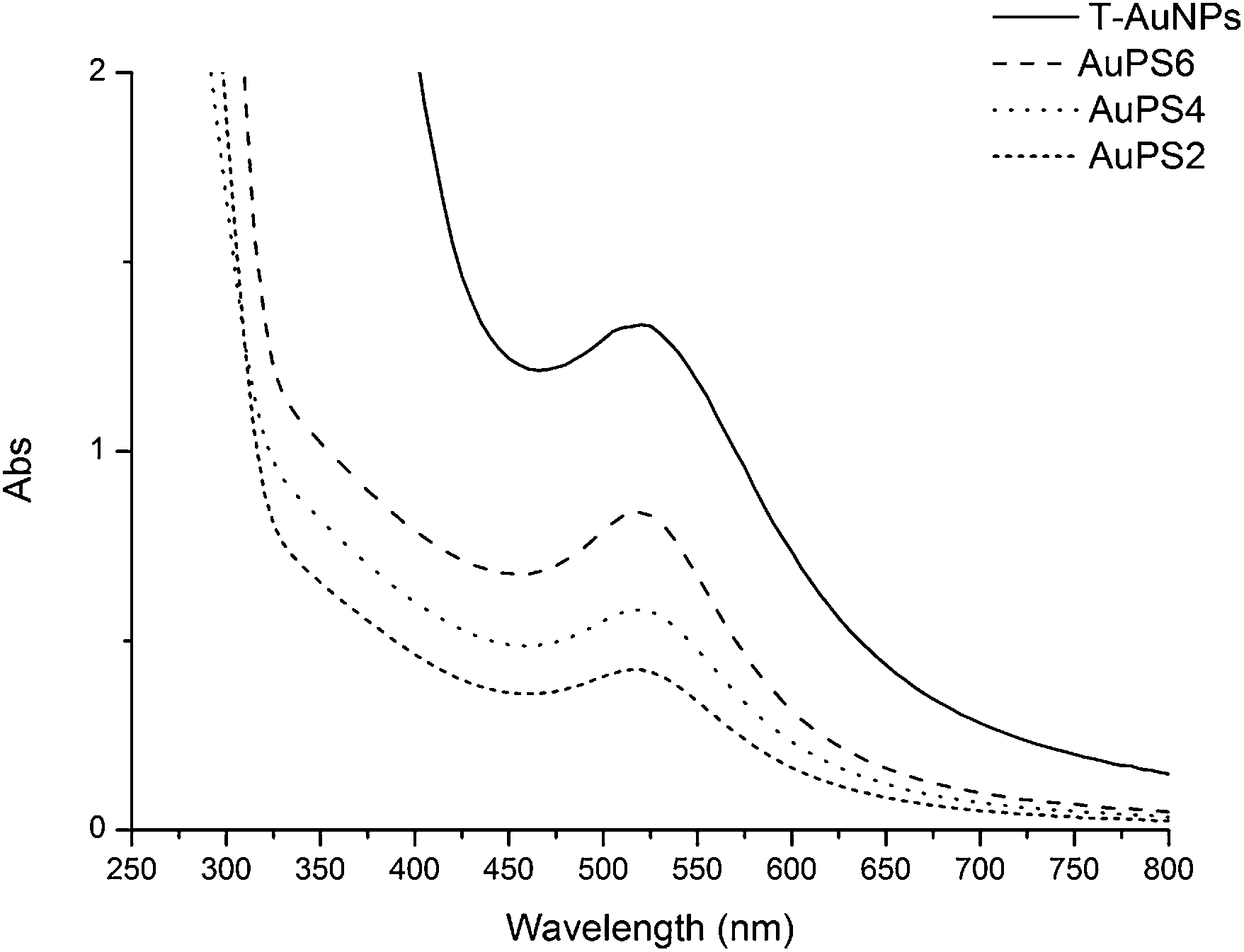 | ||
| Fig. 4 UV-Vis spectra of the gold nanoparticles coated with TEMPO and coated with polystyrene in THF solutions (T-AuNPs 0.5 mg ml−1, AuPS6 and AuPS2 1 mg ml−1, AuPS4 2 mg ml−1). | ||
TEM analyses confirmed these conclusions (Fig. 5). As we can see in Fig. 5, the obtained polystyrene coated gold nanoparticles exhibit the tendency for self-organization but it is weaker than in the case of the spin coated nanoparticles (see Fig. 1).
 | ||
| Fig. 5 TEM images of (a) AuPS2, (b) AuPS4, (c) AuPS6, scale bar 20 nm. | ||
The presence of a polymer shell on the nanogold core in the obtained nanohybrids has been proved by SEC, TG and DLS analyses. SEC analyses were carried out using both THF and DMF as eluent. The molecular data obtained for nanohybrids (AuPS), polymers cleaved from nanohybrids (CPS) and free polymers non-attached to gold surface isolated from the solution (PSF) are summarized in Table 1. The results were derived from the elugrams registered at 260 and 520 nm. The data for RID detector were consistent with the data obtained at 260 nm, therefore they have not been presented. It is worth noting that for T-AuNPs sample no peaks were observed with UV and RID detection. This indicates that TEMPO coated gold nanoparticles are completely retained by PS-DVB stationary phase and cannot be analyzed using THF as eluent. Fig. 6 presents molecular mass distribution (MMD) for AuPSs obtained from SEC analyses in THF as eluent at 520 nm detection.
| Sample | Eluent | Detection | Mn [kDa] | Mw [kDa] | PDI |
|---|---|---|---|---|---|
| a Small peak in the tailing part of main peak. | |||||
| AuPS-2 | THF | 520 nm | 51 | 56 | 1.10 |
| AuPS-4 | 51 | 57 | 1.12 | ||
| AuPS-6 | 69 | 77 | 1.11 | ||
| AuPS-2 | THF | 260 nm | 51 | 56 | 1.10 |
| AuPS-4 | 47 | 56 | 1.19 | ||
| AuPS-6 | 71 | 77 | 1.08 | ||
| CPS-2 | THF | 260 nm | 10 | 11 | 1.10 |
| CPS-4 | 10 | 11 | 1.10 | ||
| CPS-6 | 12 | 14 | 1.17 | ||
| PSF-0 | THF | 260 nm | 6 | 7 | 1.17 |
| PSF-2 | 5 | 6 | 1.20 | ||
| PSF-4 | 5 | 6 | 1.20 | ||
| PSF-6 | 11 | 13 | 1.18 | ||
| AuPS-2 | DMF extrapolated | 520 nm | 64 | 68 | 1.06 |
| AuPS-4 | 68 | 76 | 1.11 | ||
| AuPS-6 | 80 | 87 | 1.08 | ||
| T-AuNP | 21 | 31 | 1.47 | ||
| AuPS-2 | DMF/LiBr (1 g l−1) | 520 nm | 58 | 63 | 1.08 |
| AuPS-4 | 54 | 61 | 1.13 | ||
| AuPS-6 | 69 | 79 | 1.13 | ||
| T-AuNP | 4a/17 | 5a/19 | 1.25a/1.11 | ||
| AuPS-2 | DMF/LiBr (1 g l−1) | 270 nm | 59 | 63 | 1.08 |
| AuPS-4 | 57 | 61 | 1.13 | ||
| AuPS-6 | 70 | 81 | 1.15 | ||
| T-AuNP | 20 | 21 | 1.03 | ||
 | ||
| Fig. 6 Molecular mass distributions obtained from SEC analyses of polystyrene coated nanohybrids (in THF with detection at 520 nm). | ||
As we can see in Fig. 6, except for the main peak at higher masses (maximum ca. 60 kDa for AuPS2, AuPS4 and 80 kDa for AuPS6), there is also a small fraction at considerably lower masses (2.5 kDa). In our opinion this low molecular fraction contains gold nanoparticles of 1.56 nm in diameter (Fig. 1), with significantly lower polymer coverage. For smaller nanoparticles, surface coverage with TEMPO radicals is significantly lower in comparison with coverage of larger particles, thus the number of attached polystyrene chains is respectively lower. This is in line with our previous findings derived from ESR studies. A sharpened shape of small peak and significant higher retention volumes may be the result of strong interactions of unshielded polar surface of gold nanoparticles with lower coverage with PS-DVB phase.45 Bokern et al.46 have described SEC analysis of polystyren-functionalized gold nanoparticles using DMF as eluent with addition of LiBr (5g l−1). Application of this type of eluent allowed in our experiments to analyze non-polymer covered gold nanoparticles. However, we have observed that the retention time of AuPSs and T-AuNPs depends on the LiBr concentration. Linear relationships with R2 > 0.99 between molar masses and the concentration of LiBr ranged from 1 to 5 g l−1 were found, with Mn and Mw decreasing with concentration increasing. In our opinion, addition of LiBr to the mobile phase may be responsible for non-realistic underestimated values of average molecular weights. Thus, we decided to determine Mn and Mw values by the extrapolation of linear dependence to the zero concentration of LiBr in DMF. Both the extrapolated molecular masses, calculated PDIs and data obtained in DMF with 1 g l−1 LiBr (270 and 520 nm) are presented in Table 1. The extrapolated Mn and Mw are higher and in our opinion more realistic than the values obtained in THF and DMF/1 g l−1 LiBr. The excellent consistency of SEC results obtained in THF or DMF/LiBr at 520 nm (surface plasmon resonance) and 260/270 nm (absorption of aromatic rings) proves that the polystyrene chains are tethered to the surface of gold nanoparticles. The extrapolated data also show that the average molecular mass of AuPS increases with increasing time of polymerization. The MMD of T-AuNP in DMF with LiBr (1 g l−1) is bimodal, the Mw of low molecular fraction is 5 kDa. The extrapolated weight-average molar mass of the main fraction is 31 kDa. This value corresponds to ca. 150 atoms in particle. The XRD estimate of the mean particle diameter for nanocrystal gold molecules of mass of 27 kDa (137 Au atoms) gave value of 3.07 nm.47 This value is in a good agreement with the average diameter 3.62 nm of T-AuNP that can be calculated from TEM results (Fig. 1).
The relatively low PDI values allow for conclusion that the polymerization of styrene was well controlled and led to the formation of uniform chains attached to gold surface. This conclusion coincides with the data obtained for polystyrenes cleaved from AuPS (CPS samples), as evidenced by polydispersity indexes (<1.2). The average molecular weights of cleaved polystyrene (CPS samples) are nearly twice as large as PSF samples after 2 hours of polymerization. However, during the next 4 hours the polymerization of grafted chains is slower and after 6 hours Mn is similar for CPS and PSF. The presence of sacrificial mediator TEMPOL in solution is responsible for slowing of polymerization in solution in greater degree than the polymerization slowing by nitroxide radicals attached to the gold surface, thus the molecular weight of polymers in solution (PSF) is increasing slower in comparison with increasing of molecular weight of polymers reversibly attached to the gold surface (CPS).
Fig. 7 shows results of TG analyses of polystyrene coated nanohybrids (AuPS), nanoparticles used for their preparation (T-AuNPs) and free polystyrene (PSF6) isolated from the same solution as AuPS6. TGA exhibited that T-AuNPs are stable in temperature below 150 °C (in solid state under helium atmosphere). This is typical thermal stability observed for thiolate stabilized gold nanoparticles.48 The thermal decomposition of organic coating of T-AuNPs is multistage process, which consists of four main steps and leads to the solid residue (gold) constituting 56% of the total mass. The relatively low weight percentage of Au results from the presence of small particles population (see Fig. 1). The content of gold in all the obtained nanocomposites is similar and ranges from 9.3% for AuPS4 to 12.6% for AuPS2. As can be seen in Fig. 7, the polystyrene coating on the gold nanoparticles surface (AuPS) significantly improves their thermal stability. TGA curves show that the decomposition temperature of the obtained nanohybrids approached above 250 °C.
 | ||
| Fig. 7 Thermogravimetric curves of TEMPO coated (T-AuNPs), polystyrene coated gold nanoparticles (AuPS) and polystyrene non-attached to the gold surface. | ||
It is worth noting that the thermal stability of the obtained nanocomposites is slightly higher than the thermal stability of pure polystyrene. AuPS4 exhibits the highest thermal stability among studied materials.
However, the observed higher thermal stability of the obtained nanohybrids may be the result of the stability of polymer shell and does not necessarily mean the absence of aggregation process. Therefore, we decided to check properties of solid residues, after heating the obtained nanohybrids and pristine nanoparticles to appropriate temperature, in the conditions of TG measurements. For this purpose, the solid residues after TG measurements have been dissolved in THF, and next UV-Vis spectra have been recorded for the solutions obtained in this way, results for which are presented in Fig. 8. As we can see, in UV-Vis spectrum of solution, obtained as a result of dissolving solid residue after heating of T-AuNPs, in solid state to 160 °C, absorption band connected to SPR of nanogold completely disappears due to aggregation process. This is in agreement with TGA curve obtained for T-AuNPs (see Fig. 7), which shows that the thermal decomposition of T-AuNPs begins above 150 °C. The polymer shell in the synthesized nanohybrids gives them significantly higher thermal stability. SPR band in UV-Vis spectrum of solution of AuPS4 after heating to 300 °C (Fig. 8) is unchanged in comparison with SPR band in UV-Vis spectrum of THF solution of AuPS4 not subjected to heating (see Fig. 4). It means that the polymer insulating coating efficiently protects nanoparticles against aggregation even at temperature sufficient for easy breaking of Au–S bonds. Unlike only thiol protected AuNPs which are unstable at temperature above 150 °C, because their thin and thermally unstable coatings can be easily removed in those conditions, the polystyrene shells do not allow for a combination of metallic cores of the nanoparticles.
 | ||
| Fig. 8 UV-Vis spectra of THF solutions of solid residues after heating to appropriate temperature (in TG measurements conditions) polystyrene coated gold nanoparticles (AuPS4) and thiolate stabilized gold nanoparticles (T-AuNPs). | ||
TGA curves (Fig. 7) demonstrate that the thermal stability of the other obtained nanohybrids is similar.
Dynamic Light Scattering measurements (DLS) showed that the average hydrodynamic diameters of the obtained hybrid particles are higher than TEMPO coated nanoparticles used for their preparation (Fig. 9). Because TEM analyses showed that the diameters of metallic cores in the studied structures have not been changed during the polymerization process (see Fig. 5) it can be concluded that the increased size of particles is a consequence of polymer chains grafting on the gold surface of nanoparticles. The observed relationship between the sizes of nanoparticles and time of polymerization remains in very good agreement with results obtained from SEC analyses. The average molecular weights of nanohybrids after 2 h and 4 h of polymerization are very similar and increase significantly after 6 h, thus corresponding to the hydrodynamic diameters change in the same order. For AuPS4 observed tailing of the peak may be related to the presence of non-attached polystyrene in the sample (not complete purification). The presence of small quantities of additional non-attached polystyrene can explain insignificantly higher thermal stability observed in TGA in comparison with the other samples.
 | ||
| Fig. 9 The number averaged hydrodynamic diameter distribution of core–shell nanohybrids and nanoparticles used for their preparation obtained from DLS measurements. | ||
4. Conclusions
A facile and novel route to synthesis of core–shell nanohybrids, based on application of easy to obtain nitroxide covered gold nanoparticles, has been proposed. The obtained nanomaterials exhibit strong surface plasmon resonance and high thermal stability. The stability of those materials makes them possible candidates for applications as high-temperature optical sensors. Nitroxide Mediated Polymerization allowed for obtaining gold nanoparticles covered with well-defined polystyrene shell (PDI < 1.3). The proposed procedure can be easy to utilize as a tailor-made nanohybrids preparation via precisely designed macroradicals coupling.Acknowledgements
This work was supported by Project DEC-2011/01/B/ST5/03941 from National Science Centre. TEM images were obtained using the equipment purchased within CePT Project no.: POIG.02.02.00-14-024/08-00. We would like to thank Karolina Malinowska, MSc for assisting in making of TEM images and Mr Kamil Fijałkowski for helping in pictures preparation.Notes and references
- E. Bourgeat-Lami, J. Nanosci. Nanotechnol., 2002, 2, 1–24 CrossRef CAS PubMed.
- E. Bourgeat-Lami, Hybrid Materials, Synthesis, Characterization and Applications, ed. G. Kickelbick, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed and references therein.
- U. Kreibig and M. Vollmer, Optical properties of metal clusters, Springer, Berlin, 1995 Search PubMed.
- C. F. Bohren and D. R. Huffman, Absorption and scattering of light by small particles, Wiley, New York, 1983 Search PubMed.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Plasmonics, 2007, 2, 107–118 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- A. C. Templeton, J. J. Pietron, R. W. Murray and P. Mulvaney, J. Phys. Chem. B, 2000, 104, 564–570 CrossRef CAS.
- K. H. Su, Q.-H. Wei, X. Zhang, J. J. Mock, D. R. Smith and S. Schultz, Nano Lett., 2003, 3, 1087–1090 CrossRef CAS.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed and references therein.
- J. Shan and H. Tenhu, Chem. Commun., 2007, 4580–4598 RSC and references therein.
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677–710 CrossRef CAS.
- G. J. Fleer, M. A. Cohen-Stuart, J. M. H. Scheutjens, T. Cosgrove and B. Vincent, Polymers at interfaces, Chapman and Hall, London, 1993 Search PubMed.
- C. Y. Hong, Y. Z. You and C. Y. Pan, Chem. Mater., 2005, 17, 2247–2254 CrossRef CAS.
- P. Mansky, Y. Liu, E. Huang, T. P. Russell and C. J. Hawker, Science, 1997, 275, 1458–1460 CrossRef CAS.
- R. X. Chen, W. Feng, S. P. Zhu, G. Botton, B. Ong and Y. L. Wu, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1252–1262 CrossRef CAS.
- (a) D. H. Solomon, E. Rizzardo and P. Cacioli, US CSIRO 4 581 429, 1986; (b) M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2998 CrossRef CAS; (c) D. H. Solomon, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5748–5764 CrossRef CAS; (d) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed; (e) M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS PubMed; (f) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- A. Kotal, T. K. Mandal and D. Walt, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3631–3642 CrossRef CAS.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed and references therein.
- (a) J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS; (b) G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS PubMed.
- (a) A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. Mc Cormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS PubMed; (b) C. Mangeney, F. Ferrage, I. Aujard, V. Marchi-Artzner, L. Jullien, O. Ouari, El.-D. Rekai, A. Laschewsky, I. Vikholm and J. W. Sadowski, J. Am. Chem. Soc., 2002, 124, 5811–5821 CrossRef CAS PubMed; (c) M.-Q. Zhu, L.-Q. Wang, G. J. Exarhos and A. D. Q. Li, J. Am. Chem. Soc., 2004, 126, 2656–2657 CrossRef CAS PubMed; (d) M. K. Corbierre, N. S. Cameron and R. B. Lennox, Langmuir, 2004, 20, 2867–2873 CrossRef CAS; (e) S. Luo, J. Xu, Y. Zhang, S. Liu and C. Wu, J. Phys. Chem. B, 2005, 109, 22159–22166 CrossRef CAS PubMed; (f) J. Shan, M. Nuopponen, H. Jiang, T. Viitala, E. Kauppinen, K. Kontturi and H. Tenhu, Macromolecules, 2005, 38, 2918–2926 CrossRef CAS.
- (a) J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS; (b) L. Di and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959–1021 CrossRef PubMed; (c) K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- S. Nuß, H. Böttcher, H. Wurm and M. L. Hallensleben, Angew. Chem., Int. Ed., 2001, 40, 4016–4018 CrossRef.
- K. Ohno, K. Koh, Y. Tsujii and T. Fukuda, Macromolecules, 2002, 35, 8989–8993 CrossRef CAS.
- T. K. Mandal, M. S. Fleming and D. R. Walt, Nano Lett., 2002, 2, 3–7 CrossRef.
- A. Kotal, T. K. Mandal and D. R. Walt, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3631–3642 CrossRef CAS.
- D. J. Kim, S. M. Kang, B. Kong, W.-J. Kim, H. Paik, H. Choi and I. S. Choi, Macromol. Chem. Phys., 2005, 206, 1941–1946 CrossRef CAS.
- J. Raula, J. Shan, M. Nuopponen, A. Niskanen, H. Jiang, E. I. Kauppinen and H. Tenhu, Langmuir, 2003, 19, 3499–3504 CrossRef CAS.
- (a) M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS; (b) D. H. Solomon, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5748–5764 CrossRef CAS; (c) C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed; (d) J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS PubMed.
- (a) M. K. Brinks, M. Hirtz, L. F. Chi, H. Fuchs and A. Studer, Angew. Chem., Int. Ed., 2007, 46, 5231–5233 CrossRef CAS PubMed; (b) K. L. Mulfort, J. Ryu and Q. Y. Zhou, Polymer, 2003, 44, 3185–3192 CrossRef CAS; (c) L. Andruzzi, W. Senaratne, A. Hexemer, E. D. Sheets, B. Ilic, E. J. Kramer, B. Baird and C. K. Ober, Langmuir, 2005, 21, 2495–2504 CrossRef CAS PubMed; (d) L. Andruzzi, A. Hexemer, X. Li, C. K. Ober, E. J. Kramer, G. Galli, E. Chiellini and D. A. Fischer, Langmuir, 2004, 20, 10498–10506 CrossRef CAS PubMed; (e) R. Matsuno, K. Yamamoto, H. Otsuka and A. Takahara, Chem. Mater., 2003, 15, 3–5 CrossRef CAS; (f) R. Matsuno, K. Yamamoto, H. Otsuka and A. Takahara, Macromolecules, 2004, 37, 2203–2209 CrossRef CAS; (g) R. Matsuno, H. Otsuka and A. Takahara, Soft Matter, 2006, 2, 415–421 RSC; (h) S. Voccia, C. Jerome, C. Detrembleur, P. Leclere, R. Gouttebaron, M. Hecq, B. Gilbert, R. Lazzaroni and R. Jerome, Chem. Mater., 2003, 15, 923–927 CrossRef CAS; (i) K. J. Bian and M. F. Cunningham, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2145–2154 CrossRef CAS; (j) X.-D. Zhao, X.-H. Fan, X.-F. Chen, C.-P. Chai and Q.-F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4656–4667 CrossRef CAS; (k) K. Sill and T. Emrick, Chem. Mater., 2004, 16, 1240–1243 CrossRef CAS.
- O. Swiech, R. Bilewicz and E. Megiel, RSC Adv., 2013, 3, 5979–5986 RSC.
- R. Nicolay, L. Marx, P. Hemery and K. Matyjaszewski, Macromolecules, 2007, 40, 9217–9223 CrossRef CAS.
- M. Farle, M. Zomack and K. Baberschke, Surf. Sci., 1985, 160, 205–208 CrossRef CAS.
- P. G. Barkley, J. P. Hornak and J. H. Freed, J. Chem. Phys., 1986, 84, 1886–1889 CrossRef CAS PubMed.
- U. J. Katter, T. Risse, H. Schlienz, M. Beckendorf, T. Klüner, H. Hamann and H.-J. Freund, J. Magn. Res., 1997, 126, 242–247 CrossRef CAS.
- V. Chechik, H. J. Wellsted, A. Korte, B. C. Gilbert, H. Caldararu, P. Ionita and A. Caragheorgheopol, Faraday Discuss., 2004, 125, 279–291 RSC.
- E. G. Rozantzev and M. B. Neiman, Tetrahedron, 1964, 20, 131–137 CrossRef.
- R. Owenius, M. Engström, M. Lindgren and M. Huber, J. Phys. Chem. A, 2001, 105, 10967–10977 CrossRef CAS.
- R. P. Mason and J. H. Freed, J. Phys. Chem., 1974, 78, 1321–1323 CrossRef CAS.
- T. J. Stone, T. Buckman, P. L. Nordio and H. M. Mc Connell, Proc. Natl. Acad. Sci. U. S. A., 1965, 54, 1010–1017 CrossRef CAS.
- C. Vericat, M. E. Vela, G. Benitez, P. Carrob and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805–1834 RSC.
- S. Huang, G. Tsutsui, H. Sakaue, S. Shingubara and T. Takahagi, Jpn. J. Appl. Phys., 1999, 38, 1473–1476 Search PubMed.
- J. Q. Lin, H. W. Zhang, Z. Chen, Y. G. Zheng, Z. Q. Zhang and H. F. Ye, J. Phys. Chem. C, 2011, 115, 18991–18998 CAS.
- H. Yockell-Lelièvre, D. Gingras, S. Lamarre, R. Vallée and A. M. Ritcey, Plasmonics, 2013, 8, 1369–1377 CrossRef.
- G. Wei and F. Liu, J. Chromatogr. A, 1999, 836, 253–260 CrossRef CAS.
- S. Bokern, K. Gries, H. H. Görtz, V. Warzelhan, S. Agarwal and A. Greiner, Adv. Funct. Mater., 2011, 21, 3753–3759 CrossRef CAS.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433 CrossRef CAS.
- S. Chen and K. Kimura, Langmuir, 1999, 15, 1075–1082 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Results of elemental analysis, FT-IR spectra, MS-ES+ spectrum of DiSS, ESR spectrum of DiSS, photographs of selected samples, EDS analysis results. See DOI: 10.1039/c4ra01770f |
| This journal is © The Royal Society of Chemistry 2014 |