DOI:
10.1039/C4RA01767F
(Paper)
RSC Adv., 2014,
4, 25532-25539
Novel electro-optic chromophores based on substituted benzo[1,2-b:4,5-b′]dithiophene π-conjugated bridges†
Received
28th February 2014
, Accepted 11th April 2014
First published on 16th April 2014
Abstract
Two novel non-linear optical (NLO) chromophores were designed and synthesized based on the substituted benzo[1,2-b:4,5-b′]dithiophene unit (BDT), tricyanofuran (TCF) electron acceptor and two different electron donors. These new chromophores, which exhibited good thermal stability and solubility in common organic solvents, were systematically characterized by thermogravimetric analysis, UV-Vis spectra, density functional theory (DFT) calculations and measurements of the electro-optic (EO) coefficients. Compared with the dodecyl group in chromophore BDT1 that we have previously reported, the isooctane group in BDT2 could act as a suitable isolation group, and as a result, the guest–host system containing 30% of BDT2 in amorphous polycarbonate (APC) displayed the largest EO coefficient of 102 pm V−1, which was greatly improved over BDT1 and the analogous thiophene-based chromophore with the same electron donor and acceptor. The solvatochromic analysis and DFT study demonstrated that the julolidine group in BDT3 possessed a stronger donating ability. However, the 30% BDT3/APC exhibited an EO coefficient of only 82 pm V−1, which was probably caused by the diminishment of the hyperpolarizability value of BDT3 in the polar polymer matrix. These results indicate the potential of the isooctane-substituted benzo[1,2-b:4,5-b′]dithiophene π-conjugated bridge in chromophore design and NLO materials, and further illustrate the critical role of suitable isolation groups and fine-tuning of the donor's electron-donating strength in optimizing the non-linear properties of NLO chromophores.
Introduction
With the rocketing development of information technology, electro-optic (EO) materials, which are widely used in telecommunications such as phased array radar, optical gyroscopes and modulators, have drawn great attention in the past decades.1–10 Compared to inorganic/semi-conductor EO materials, organic EO materials have many advantages such as large non-linear optical (NLO) coefficient, ultra-fast response time, large bandwidth, low dielectric constant and good processability.2,11,12 In order to promote the development of EO materials, much effort has been expended in developing NLO chromophores and EO polymers. But to fulfil the requirements of device fabrication and operation, a strong need remains for improving the NLO properties and stability of the NLO chromophores, which play a key role in EO devices at both the microscopic level and macroscopic materials level.13–15
Organic second-order NLO chromophore molecules are the core component of EO materials and typically consist of a π-conjugated bridge end-capped with strong electron-donating and -accepting groups.2,11 Among them, a π-conjugated electron bridge helping intramolecular charge-transfer plays an essential role in the chromophore structure.11,14,16 For the purpose of improving microscopic first-order hyperpolarizability (β), increasing and optimizing the π-conjugated bridge is a critical aspect. But unfortunately, the added conjugation length often influences the stability of the molecule, which will directly decrease the practical application of NLO chromophoresc.2,11 The benzo[1,2-b:4,5-b′]dithiophene (BDT) unit containing two thienyl rings has been widely used for photovoltaic conjugated polymers due to its large planar conjugated structure, which makes BDT a promising electron bridge.17–19 Previously, we first introduced this unit into NLO materials and synthesized a novel chromophore BDT1 which showed promising second-order NLO features and could meet the requirement of stability for application.20 However, the guest–host EO materials21,22 of BDT1 doped into amorphous polycarbonate (APC) did not exhibit the desired EO activities.
Suppressing the dipolar–dipolar electrostatic interaction and increasing the microscopic hyperpolarizability are basic strategies to improve the EO activities from the angle of NLO chromophores. Dipolar–dipolar electrostatic interaction between chromophores, which could cause aggregation of the chromophores, is a major obstacle hindering translation of the microscopic hyperpolarizability into macroscopic EO activity.5,23–30 Work from the groups of Dalton et al. and Li et al. has demonstrated that the dipole–dipole interactions could be effectively suppressed through the introduction of suitable isolation groups.31–35 And the relative researches also predicted that the ideal shape for the chromophore is spherical.2,36 On the other hand, the hyperpolarizability is the source of EO activity, so improving the chromophores' NLO properties is also an effective approach to improve macroscopic activity, and the hyperpolarizability can be tuned by optimizing the ground-state polarization through modification of the strength of the donor or acceptor.
To improve the macroscopic EO activities of BDT-based NLO chromophores and further explore the potential of the BDT π-conjugated bridge in NLO materials, we designed and synthesized the novel chromophore BDT2 with new isolation groups and BDT3 with a stronger electron donor as shown in Chart 1. BDT2 adopts the shorter dendritic isooctane groups, which we believe are more suitable for isolating and can make the chromophore closer to a spherical shape. In BDT3, the julolidine derivative 8-hydroxy-1,1,7,7-tetramethyl-formyljulolidine, which has been proved to be an excellent electron donor,13,37 is used to improve the electron-donating ability. The thermal stability, photophysical properties, density functional theory (DFT) calculations and EO activities of these chromophores were systematically studied and compared. The results further proved that substituted benzo[1,2-b:4,5-b′]dithiophene is a quite promising π-conjugated bridge in chromophore design and that the isooctane group could be a suitable isolation group to improve the EO activities (maximum r33 value was up to 102 pm V−1), and they also illustrated the critical role of fine-tuning of donor's electron-donating strength in optimizing the non-linear properties of NLO chromophores.
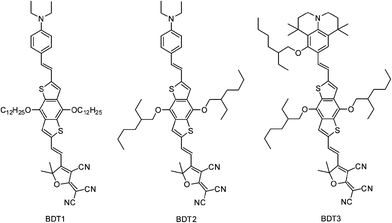 |
| | Chart 1 Chemical structures of the chromophores. | |
Experimental
Materials and instruments
All reagents were purchased from commercial sources and were used as received unless stated. DMF, THF, diethyl ether and acetone were freshly distilled prior to use. Compound 3 was synthesized by our group, 4,8-dihydrobenzo[1,2-b:4,5-b′]dithiophen-4,8-dione and the TCF acceptor were prepared according to the literature.18,38–401H NMR and 13C NMR spectra were measured using an AVANCE 400 spectrometer (Bruker) using tetramethylsilane (TMS; δ = 0 ppm) as the internal standard (resolution 1H ≤ 0.45 Hz; 13C ≤ 0.2 Hz). High-resolution mass spectrometry experiments were performed using a Bruker Daltonics Apex IV FTMS spectrometer (accuracy < 1–2 ppm). The MS spectra were obtained using MALDI-TOF (Matrix Assisted Laser Desorption/Ionization of Flight) on a GCT Premier (Waters) (resolution 7000 FWHM; sensitivity 1 pg). Melting points were obtained using a Beijing X4 melting point tester. Fourier transform infrared (FT-IR) spectra were recorded using a Varian 3100 FT-IR spectrometer (resolution 0.20 cm−1; accuracy < 0.01 cm−1). UV-Vis spectra were obtained using a Varian Cary5000 spectrophotometer (resolution 0.05 nm; accuracy ≤ ±0.1 nm). The TGA curve was recorded using a TA-instrument Q50 analyzer with a heating rate of 10 °C min−1 under a nitrogen atmosphere.
Syntheses
Synthesis of compound 1.
4,8-Dihydrobenzo[1,2-b:4,5-b′]dithiophen-4,8-dione (4.4 g, 20 mmol), zinc powder (3.3 g, 50 mmol), and water (60 mL) were put into a 250 mL flask, then NaOH (12 g, 0.3 mol) was added into the mixture. After the mixture was stirred and refluxed for 1 h, 1-bromoisooctane (11.6 g, 60 mmol) and a catalytic amount of tetrabutylammonium bromide were added into the flask. The reactant was refluxed for 8 h and then was poured into cold water and extracted by diethyl ether three times. The organic layer was dried over anhydrous MgSO4. After the solvent had been removed, the crude product was purified by column chromatography. Compound 1 (7.1 g, yield 79%) was obtained as a pale yellow oil. IR (KBr), νmax cm−1: 3084, 2959, 2927, 2872 (C–H), 1519 (thienyl), 1439 (benzene ring), 1198 (Ph–O–C). MS, m/z: 446.23 (M+). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.50 (thienyl, d, J = 5.5 Hz, 2H), 7.37 (thienyl, d, J = 5.5 Hz, 2H), 4.26–4.15 (–O–CH2–, m, 4H), 1.83 (–CH–, m, J = 11.9, 5.8 Hz, 2H), 1.77–1.35 (–CH2–, m, 16H), 1.00 (–CH3, m, 12H).
Synthesis of compound 2.
Compound 1 (2.23 g, 5 mmol) was dissolved in dry THF (30 mL) in a 100 mL flask. Then n-butyllithium (3.75 mL, 15 mmol, 2.5 M) was injected into the solution slowly at −78 °C under an inert atmosphere. The solution was stirred for 30 min at −78 °C and for 30 min at −10 °C. After that, the solution was cooled at −78 °C and excess DMF was injected into the solution slowly. The solution was stirred at −78 °C for 30 min and then warmed to room temperature. After that the reactant was quenched with water (300 mL), extracted with diethyl ether, and dried over anhydrous MgSO4. The crude product was purified by column chromatography. Compound 2 (1.80 g, yield 72%) was obtained as an orange solid. Mp: 93–94 °C. IR (KBr),νmax cm−1: 2960, 2930, 2874 (C–H), 1670 (–CHO), 1535, 1452 (aromatic ring), 1215 (Ph–O–C). MS, m/z: 502.43 (M+). 1H NMR (400 MHz, CDCl3) δ (ppm) 10.14 (–CHO, s, 2H), 8.18 (thienyl, s, 2H), 4.28 (–O–CH2–, d, J = 5.3 Hz, 4H), 1.91–1.80 (–CH–, m, 2H), 1.74–1.34 (–CH2–, m, 16H), 0.98 (–CH3, m, 12H).
Synthesis of compound 4.
Compound 2 (1.10 g, 2.2 mmol), compound 3 (1.01 g, 2 mmol), NaH (1.0 g, 0.04 mol), and dry diethyl ether (50 mL) were put into a 100 mL flask. The reactant was stirred for 24 h at ambient temperature with the flask sealed up. After that, the mixture was poured into ice water (200 mL) slowly. The mixture was then extracted with diethyl ether and dried over MgSO4. After removal of the diethyl ether, the crude product was purified by column chromatography. Compound 4 (0.82 g, yield 58%) was obtained as a red oil. 1H NMR (CDCl3) shows that the product is a mixture of trans and cis isomers. On the basis of the integration areas of two –CHO signals, the trans isomer is estimated to be 90% and the cis isomer 10%. IR (KBr), νmax cm−1: 2959, 2926, 2870 (C–H), 1670 (–CHO), 1599, 1520, 1449 (aromatic ring), 1265 (Ph–O–C). MS, m/z: 647.12 (M+). 1H NMR (400 MHz, CDCl3) δ (ppm) 10.11 (–CHO, s, 0.10H), 10.06 (–CHO, s, 0.90H), 8.15 (thienyl-H, s, 0.10H), 8.11 (thienyl, s, 0.90H), 7.39 (phenyl, d, J = 8.7 Hz, 2H), 7.24 (thienyl, s, 1H), 7.09 (vinylic, d, J = 15.9 Hz, 1H), 6.95 (vinylic, d, J = 15.9 Hz, 1H), 6.66 (phenyl, d, J = 8.7 Hz, 2H), 4.36–4.09 (–O–CH2–, m, 4H), 3.40 (–N–CH2–, q, J = 7.0 Hz, 4H), 1.88–1.76 (–CH–, m, 2H), 1.69–1.39 (–CH2–, m, 16H), 1.19 (–CH3, t, J = 7.0 Hz, 6H), 1.03 (–CH3, t, J = 7.6 Hz, 6H), 0.99–0.94 (–CH3, m, 6H).
Synthesis of compound 5.
8-Hydroxy-1,1,7,7-tetramethyl-formyljulolidine (2.73 g, 0.01 mol), 1-bromoisooctane (2.90 g, 0.015 mol) and K2CO3 (3.0 g, 0.022 mol) were added into anhydrous acetone (50 mL). The mixture was stirred and refluxed for 15 h. After the precipitates were filtered out and the solvent was removed, the residue was purified by column chromatography. Compound 5 was gained as a pale yellow oil (3.03 g, yield 81%). IR (KBr), νmax cm−1: 2957, 2930, 2857 (C–H), 1661 (–CHO), 1589, 1516 (benzene ring), 1233 (Ph–O–C). MS, m/z: 385.59 (M+). 1H NMR (400 MHz, CDCl3) δ (ppm) 9.98 (–CHO, s, 1H), 7.62 (phenyl, s, 1H), 3.90–3.80 (–O–CH2–, m, 2H), 3.32–3.26 (–N–CH2–, m, 2H), 3.25–3.19 (–N–CH2–, m, 2H), 2.00–1.89 (–CH–, m, 1H), 1.76–1.65 (–NCH2–CH2–, m, 4H), 1.61–1.19 (–CH2– and –CH3, m, 20H), 0.92 (–CH3, m, 6H).
Synthesis of compound 7.
Compound 5 (3.85 g, 0.010 mol) was dissolved into methanol (40 mL) and stirred well, and then sodium borohydride (0.5 g, 0.013 mol) was added gradually at 0 °C. Then the reactant was stirred for 24 h at ambient temperature. After that, the methanol was removed and water was added. The aqueous solution was neutralized with dilute hydrochloric acid and extracted with dichloromethane. The combined organic layer was dried over MgSO4. Dichloromethane was removed under vacuum to get a pale yellow oil. The oil was dissolved in chloroform (40 mL) and triphenylphosphine hydrobromide (3.10 g, 0.009 mol) was added. The reactant was refluxed for 3 h and then the chloroform was removed. The precipitates were washed several times with diethyl ether to gain compound 6 as a white powder directly for the next step. Compound 7 was prepared from compound 2 and compound 6 according to the synthesis of compound 4. Compound 7 was obtained as a red oil (yield 54%). 1H NMR (CDCl3) shows that the product is a mixture of trans and cis isomers. On the basis of the integration areas of two thienyl-H signals, the trans isomer is estimated to be 82% and the cis isomer 18%. IR (KBr), νmax cm−1: 2957, 2926, 2858 (C–H), 1674 (–CHO), 1504, 1456 (aromatic ring), 1250 (Ph–O–C). MS, m/z: 855.75 (M+). 1H NMR (400 MHz, CDCl3) δ (ppm) 10.00 (–CHO, s, 1H), 8.08 (thienyl, s, 0.18H), 8.05 (thienyl, s, 0.82H), 7.15 (thienyl, phenyl and vinylic, m, 3H), 6.99 (vinylic, d, J = 15.9 Hz, 1H), 4.25–4.01 (–O–CH2–, m, 4H), 3.78–3.61 (–O–CH2–, m, 2H), 3.15–2.99 (–N–CH2–, m, 4H), 2.00–1.87 (–CH–, m, 1H), 1.76 (–CH–, m, 2H), 1.70–1.09 (–CH2– and –CH3, m, 40H), 0.94 (–CH3, t, J = 8.2 Hz, 6H), 0.89–0.84 (–CH3, m, 6H), 0.77 (–CH3, t, J = 6.9 Hz, 6H).
Synthesis of chromophore BDT2.
Compound 4 (0.33g, 0.5 mmol) was dissolved in chloroform (20 mL), and then a catalytic account of triethylamine and TCF (0.14 g, 0.75 mmol) were added. The mixture was stirred and refluxed for 5 h. After the solvent had been removed, the crude product was purified by column chromatography. Chromophore BDT2 (0.23 g, yield 56%) was obtained as a dark blue solid. Mp: 224–225 °C. IR (KBr), νmax cm−1: 2960, 2928, 2870 (C–H), 2227 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1601, 1539 (aromatic ring), 1285 (Ph–O–C). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.98 (vinylic, d, J = 15.8 Hz, 1H), 7.81 (thienyl, s, 1H), 7.77 (phenyl, d, J = 8.2 Hz, 2H), 7.68 (phenyl, d, J = 8.2 Hz, 2H), 7.46 (thienyl, s, 1H), 7.41 (vinylic, d, J = 16.0 Hz, 1H), 7.03 (vinylic, d, J = 16.0 Hz, 1H), 6.67 (vinylic, d, J = 15.8 Hz, 1H), 4.27 (–O–CH2–, d, J = 5.6 Hz, 2H), 4.19 (–O–CH2–, d, J = 5.5 Hz, 2H), 3.67 (–N–CH2–, s, 2H), 3.33 (–N–CH2–, s, 2H), 1.88–1.76 (–CH–, m, 2H), 1.81 (–CH3, s, 6H), 1.77–1.22 (–CH2– and –CH3, m, 22H), 1.04 (–CH3, t, J = 7.4 Hz, 6H), 0.96 (–CH3, t, J = 6.5 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ (ppm) 175.43, 172.31, 148.46, 147.07, 146.25, 143.37, 140.15, 138.50, 136.55, 133.61, 131.15, 130.80, 130.52, 128.65, 128.27, 123.04, 117.73, 116.64, 114.76, 112.04, 111.82, 111.34, 110.85, 97.98, 97.29, 57.12, 44.58, 40.81, 30.44, 29.30, 26.47, 23.94, 23.30, 14.37, 12.85, 11.48. HRMS: for C50H61N4O3S2, calcd: 829.41796, found: 829.41653.
N), 1601, 1539 (aromatic ring), 1285 (Ph–O–C). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.98 (vinylic, d, J = 15.8 Hz, 1H), 7.81 (thienyl, s, 1H), 7.77 (phenyl, d, J = 8.2 Hz, 2H), 7.68 (phenyl, d, J = 8.2 Hz, 2H), 7.46 (thienyl, s, 1H), 7.41 (vinylic, d, J = 16.0 Hz, 1H), 7.03 (vinylic, d, J = 16.0 Hz, 1H), 6.67 (vinylic, d, J = 15.8 Hz, 1H), 4.27 (–O–CH2–, d, J = 5.6 Hz, 2H), 4.19 (–O–CH2–, d, J = 5.5 Hz, 2H), 3.67 (–N–CH2–, s, 2H), 3.33 (–N–CH2–, s, 2H), 1.88–1.76 (–CH–, m, 2H), 1.81 (–CH3, s, 6H), 1.77–1.22 (–CH2– and –CH3, m, 22H), 1.04 (–CH3, t, J = 7.4 Hz, 6H), 0.96 (–CH3, t, J = 6.5 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ (ppm) 175.43, 172.31, 148.46, 147.07, 146.25, 143.37, 140.15, 138.50, 136.55, 133.61, 131.15, 130.80, 130.52, 128.65, 128.27, 123.04, 117.73, 116.64, 114.76, 112.04, 111.82, 111.34, 110.85, 97.98, 97.29, 57.12, 44.58, 40.81, 30.44, 29.30, 26.47, 23.94, 23.30, 14.37, 12.85, 11.48. HRMS: for C50H61N4O3S2, calcd: 829.41796, found: 829.41653.
Synthesis of chromophore BDT3.
BDT3 was prepared from compound 7 and TCF according to the synthesis of BDT2. BDT3 was obtained as a dark green solid (yield 48%). 1H NMR (acetone) of the product shows two sets of similar signals, indicating that the product is a mixture of trans and cis isomers. On the basis of the integration areas of two thienyl-H signals, the trans isomer is estimated to be 55% and the cis isomer 45%. Mp: 103–105 °C. IR (KBr), νmax cm−1: 2957, 2930, 2870 (C–H), 2228 (CN), 1539, 1479 (aromatic ring), 1288 (Ph–O–C). 1H NMR (400 MHz, acetone) δ (ppm) 8.32 (vinylic, d, J = 16.1 Hz, 1H), 8.20 (thienyl, s, 0.45H), 8.19 (thienyl, s, 0.55H), 7.58 (vinylic, d, J = 13.4 Hz, 0.45H), 7.51 (thienyl, s, 0.55H), 7.43 (phenyl, s, 1H), 7.49 (thienyl, s, 0.45H), 7.37 (vinylic, d, J = 16.1 Hz, 0.55H), 7.24 (vinylic, d, J = 16.2Hz, 1H), 6.96 (vinylic, d, J = 16.0, 1H), 4.42–4.17 (–O–CH2–, m, 4H), 3.81 (–O–CH2–, dd, J = 18.6, 7.6 Hz, 2H), 3.28–3.15 (–O–CH2–, m, 4H), 1.94 (–CH3, s, 6H), 1.91–1.81 (–CH–, m, 3H), 1.80–1.25 (–CH2– and –CH3, m, 40H), 1.05 (–CH3, t, J = 7.3 Hz, 6H), 0.97–0.93 (–CH3, m, 6H), 0.89–0.85 (–CH3, m, 6H). HRMS: for C64H84N4O4S2, calcd: 1036.59285, found: 1036.59080.
Film preparation
For r33 measurements, guest–host polymer films were prepared by doping the chromophores into amorphous polycarbonate (APC; Tg = 190 °C). The chromophores (20–30 mg) were mixed with APC (100 mg) in dibromomethane (1.0–1.1 mL) as the solvent. Stirred for 12 h, the solutions were filtered through a 0.22 μm Teflon membrane filter and spin-coated onto indium tin oxide (ITO) glass substrates at room temperature with a spinning rate of 600 rpm to produce the films, which were then baked overnight at room temperature under vacuum, yielding thin films of optical quality and a thickness of around 3 μm. The poling process was carried out at a temperature of about 10 °C above the Tg of the polymers.
Results and discussion
Synthesis and characterization
The synthetic approach for the new chromophores BDT2 and BDT3 based on substituted BDT π-conjugated bridges is depicted in Scheme 1. 4,8-Dihydrobenzo[1,2-b:4,5-b′]dithiophen-4,8-dione reacted with 1-bromoisooctane after reduction by zinc and sodium hydroxide to obtain compound 1. Compound 2 was obtained through formylation of compound 1 with n-BuLi and DMF. 8-Hydroxy-1,1,7,7-tetramethyl-formyljulolidine was alkylated with 1-bromoisooctane in the presence of potassium carbonate to obtain compound 5. Both donors were reduced by sodium borohydride followed by a reaction with triphenylphosphonium bromide to form Wittig salts, which reacted with compound 2, finally followed a Knoevenagel condensation with the TCF acceptor to obtain the target chromophores. The structures of the chromophores were confirmed by 1H NMR, HRMS, and UV-Vis spectroscopic analysis, and the data obtained were in full agreement with the proposed formulations.
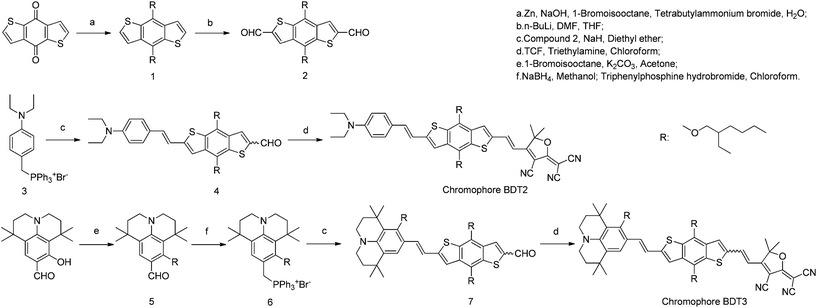 |
| | Scheme 1 Synthesis route of BDT2 and BDT3. | |
Physical properties of the chromophores
Thermal stability
Thermal stability is very important for application of the chromophores in NLO devices because most processes used to prepare NLO devices are carried out under high temperature: the temperature should be above the glass transition temperature of the NLO polymers in the poling process; high temperature is needed to drive the solvent away in the process of film preparation (including cladding and coring films); and higher temperature would be needed in the preparation process of the electrode. To investigate the thermal stability of the chromophores, thermogravimetric analysis (TGA) was used. TGA curves of BDT1, BDT2, and BDT3 are shown in Fig. 1 and the thermal decomposition temperature (Td, 5% lost) values obtained from TGA were 226, 294 and 271 °C, respectively. It is easy to see that shortening of the flexible chains in the BDT unit can significantly improve the thermal stability of this kind of chromophore to meet the higher requirements of application. We believe that it is the overlong side chains that induce the instability of the conjugated structure of BDT1.
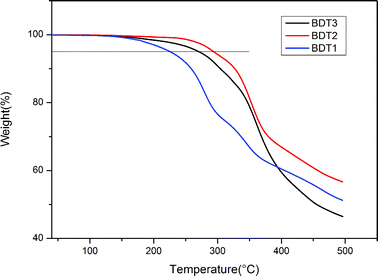 |
| | Fig. 1 Thermogravimetric analyses of the chromophores. | |
Optical properties
In order to reveal the distinctions of the electronic structures of the chromophores, UV-Vis absorption spectra were measured in a series of aprotic solvents with different polarities so that the solvatochromic behavior of each chromophore could be investigated in a wide range of dielectric environments. The UV-Vis absorption spectra of the chromophores in six different solvents are presented in Fig. 2, and the data are summarized in Table 1.The three chromophores in all solvents all exhibited a quite similar broad π–π* intramolecular charge-transfer absorption band. Also, they all showed a bathochromic shift of its absorption maximum initially and then reversed to a hypsochromic shift for more polar solvents like dichloromethane, acetone and acetonitrile, namely inverted solvatochromism. This indicated that these three chromophores were all quite dipolar. Compared with the spectra of BDT1 and BDT2, which were almost the same due to their same conjugated structure, the spectra of BDT3 exhibited an obvious red-shift. The maximal absorption wavelength (λmax) values of BDT1, BDT2, and BDT3 in chloroform were 655, 659 and 672 nm, respectively. Compared to BDT2, the λmax of BDT3 red-shifted 13 nm owing to its stronger electron donor. Δλmax (the difference in dioxane and in chloroform) values of BDT1, BDT2 and BDT3 are 54, 58 and 83 nm, respectively. According to ref. 14, larger solvatochromic effects mean a larger energy shift between different solvents and usually implied a better electro-optic performance.
 |
| | Fig. 2 UV-Vis absorption spectra of chromophores (a) BDT1, (b) BDT2 and (c) BDT3 in six different solvents (10−5 M) of varying dielectric constants (1,4-dixoane: 2.25; toluene: 2.38; chloroform: 4.81; dichloromethane: 8.93; acetone: 20.7; acetonitrile: 37.5) at room temperature and (d) UV-Vis absorption spectra of chromophores in chloroform. | |
Table 1 UV-Vis absorption and solvatochromic data of the chromophores
| |
λ
max
a/nm |
λ
max
b/nm |
λ
max
c/nm |
λ
max
d/nm |
λ
max
e/nm |
λ
max
f/nm |
Δλmaxg/nm |
|
λ
max measured in dioxane.
λ
max measured in toluene.
λ
max measured in chloroform.
λ
max measured in dichloromethane.
λ
max measured in acetone.
λ
max measured in acetonitrile.
Δλmax was the difference between λmaxa and λmaxc.
|
| BDT1 |
601 |
625 |
655 |
640 |
595 |
595 |
54 |
| BDT2 |
601 |
625 |
659 |
641 |
595 |
595 |
58 |
| BDT3 |
589 |
635 |
672 |
653 |
599 |
599 |
83 |
Theoretical calculations
For further comparison, we have performed the quantum chemical calculations within a framework of GAUSSIAN03 using the split valence B3LYP 6-31G (d,p) basis set.41–43 The density functional theory (DFT) calculations were carried out to show the electron density of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of the chromophores and the β value44 of the chromophores in a vacuum. The theoretical calculation of BDT3 was based on the trans isomer. The data obtained from our DFT calculations are summarized in Fig. 3 and Table 2.
 |
| | Fig. 3 HOMO, LUMO energy level, HOMO–LUMO energy gap (ΔE) and hyperpolarizability (β) of the chromophores using DFT calculations. | |
Table 2 Molecular orbital composition (%) in the ground state for the chromophores
| |
BDT1 |
BDT2 |
BDT3 |
| HOMO |
LUMO |
HOMO |
LUMO |
HOMO |
LUMO |
| Donor |
56.4 |
2.2 |
56.6 |
2.2 |
65.7 |
2.8 |
| π bridge |
39.9 |
50.3 |
39.7 |
50.4 |
31.5 |
50.0 |
| Acceptor |
3.7 |
47.5 |
3.7 |
47.4 |
2.8 |
47.2 |
In Fig. 3, it is easy to see that the electrons of the HOMO are localized predominantly at the donor and electron-bridge, while in the excited state those of the LUMO are localized predominantly at the acceptor and electron-bridge, which indicated that these chromophores had good intramolecular charge-transfer ability and that BDT was an excellent electron-bridge. The compositions of the HOMOs and LUMOs of these chromophores in the ground state were also calculated using the Multiwfn program with Rose Schuit (SCPA) partition45 and listed in Table 2. The molecular orbital compositions of BDT1 and BDT2 were nearly the same whereas the electron donor of BDT3 contributed a lot more to the composition of the HOMO, which prominently showed that the julolidine moiety possesses a stronger electron-donating ability compared to the aniline moiety. The calculated ΔE (HOMO–LUMO energy gap) of the chromophores also confirmed the results. And as we expected, the β values of BDT1 (2079.6 × 10−30 esu) and BDT2 (2085.2 × 10−30 esu) are very close whereas that of BDT3 (2303.1 × 10−30 esu) is much larger because BDT1 and BDT2 have the same conjugated structure while BDT3 has a stronger electron donor.
Non-inear optical property
For application, the large hyperpolarizability has to be translated into a large EO coefficient (r33). Guest–host EO polymers based on APC as the hosts were prepared to investigate the macroscopic NLO property of these chromophores. The corona poling process was carried out at a temperature about 10 °C above the Tg of the polymers. The r33 values of the poled films were measured using the Teng–Man simple reflection technique at the wavelength of 1.31 μm.46 The r33 values were calculated by the following equation:
where r33 is the EO coefficient of the poled polymer, λ is the optical wavelength, θ is the incidence angle, Ic is the output beam intensity, Im is the amplitude of the modulation, Vm is the modulating voltage, and n is the refractive index of the polymer film.
The EO coefficients of the EO poled films with loading densities (chromophores into APC) of 20 and 30 wt% were measured and are listed in Table 3. With the same loading density, the r33 values of the poled EO films doped with BDT2 were the largest, with BDT3 came second, and with BDT1 were the lowest. Compared with BDT1, the r33 of BDT2 increased over 50% with the alteration of the dodecane groups. That is because the dendritic isooctane groups can increase the steric hindrance between the chromophores to suppress the intermolecular dipolar–dipolar electrostatic interaction. And the shortening of the chains on BDT probably makes the chromophore easier to move during the poling process as well. The maximum r33 of 102 pm V−1 was obtained from 30 wt% BDT2/APC. Compared to an analogous thiophene-based FTC-type chromophore (20–50 pm V−1), the r33 increase was more than double and can absolutely meet the requirement. In BDT3, the introduction of the stronger electron donor obviously increased the hyperpolarizability, but unexpectedly, the films doped with BDT3 with the maximum β value did not achieve the maximum r33 value. Jen et al. once reported that the electro-optic polymer based on poly(methyl methacrylate) and the isophorone-derived tetraene chromophore with julolidine as the donor obtained a low EO coefficient because the hyperpolarizability of the chromophore decreased much more obviously than similar chromophores based on the aniline donor in a polar medium.6 This could also explained well why films doped with BDT3 obtained a lower EO coefficient, which was probably owing to the diminishment of the hyperpolarizability value of BDT3 in the polar polymer matrix, and these results illustrate the critical role of fine-tuning of donor's electron-donating strength in optimizing the non-linear properties of NLO chromophores.
Table 3 EO coefficients (pm V−1) of chromophores doped in APC with different loading densities
| Loading density (wt%) |
BDT1 |
BDT2 |
BDT3 |
| 20 |
57 |
87 |
68 |
| 30 |
66 |
102 |
82 |
Conclusions
Two novel chromophores BDT2 and BDT3 were designed and synthesized through modification of BDT1 which we reported previously. These chromophores were all very soluble in common solvents and were systematically characterized by MS, NMR and UV-Vis absorption spectra. Compared with BDT1 with overlong side chains that induce the instability of the conjugated structure of BDT1, the thermal stabilities of BDT2 and BDT3 were both improved and could meet the higher requirements for application. Meanwhile, DFT calculations were carried out to analyze the chromophores. Because BDT1 and BDT2 have the same conjugated structure, their UV-Vis absorption spectra, HOMO–LUMO gap and calculated first-order hyperpolarizability were nearly the same. BDT3 showed stronger bathochromic behavior in the UV-Vis absorption spectra, and owned a smaller energy gap and higher first-order hyperpolarizability in a vacuum due to its stronger electron-donor julolidine moiety. EO polymers were prepared by the chromophores being doped into APC and their r33 values were measured. The r33 values of EO polymers based on BDT2 and BDT3 were larger than the ones based on BDT1 due to the variation of the flexible chains on the BDT unit because the dendritic isooctane groups are more suitable isolation groups than dodecyl groups and can suppress dipolar–dipolar electrostatic interactions more effectively. The materials based on BDT3 with the maximum β value in a vacuum did not achieve the maximum r33 value, probably due to the diminishment of the β value in the polar polymer matrix. The maximum r33 of 102 pm V−1 was obtained from 30 wt% BDT2/APC. Compared to an analogous thiophene-based FTC-type chromophore, the r33 increase was more than double. The substituents on the BDT unit significantly influence the properties of this type of chromophore and the dendritic isooctane group is a quite suitable isolation group. These results indicate the importance of suitable isolation groups and fine-tuning of the donor's electron-donating strength in optimizing the non-linear properties of NLO chromophores. The BDT unit with suitable isolation groups can be very helpful in chromophore design and is promising in NLO materials.
Acknowledgements
We are grateful to the Directional Program of the Chinese Academy of Sciences (KJCX2.YW.H02), Innovation Fund of Chinese Academy of Sciences (CXJJ-11-M035) and National Natural Science Foundation of China (no. 11104284 and no. 61101054) for the financial support.
Notes and references
- P. A. Sullivan and L. R. Dalton, Acc. Chem. Res., 2010, 43, 10–18 CrossRef CAS PubMed.
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55 CrossRef CAS PubMed.
-
L. R. Dalton, P. A. Sullivan, D. Bale, B. Olbricht, J. Davies, S. Benight, I. Kosilkin, B. H. Robinson, B. E. Eichinger and A. K.-Y. Jen, in Organic Thin Films for Photonic Applications, ed. W. N. Herman, S. R. Flom and S. H. Foulger, American Chemical Society, 2010, vol. 1039, pp. 13–33 Search PubMed.
- H. Huang, G. Deng, J. Liu, J. Wu, P. Si, H. Xu, S. Bo, L. Qiu, Z. Zhen and X. Liu, Dyes Pigm., 2013, 99, 753–758 CrossRef CAS PubMed.
- W. Wu, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, Chem. – Eur. J., 2013, 19, 630–641 CrossRef CAS PubMed.
- X.-H. Zhou, J. Luo, J. A. Davies, S. Huang and A. K. Y. Jen, J. Mater. Chem., 2012, 22, 16390 RSC.
- C. Coluccini, A. K. Sharma, M. Caricato, A. Sironi, E. Cariati, S. Righetto, E. Tordin, C. Botta, A. Forni and D. Pasini, Phys. Chem. Chem. Phys., 2013, 15, 1666 RSC.
- C. Coluccini, P. Metrangolo, M. Parachini, D. Pasini, G. Resnati and P. Righetti, J. Mater. Sci.: Mater. Electron., 2008, 46, 5202–5213 CAS.
- I. Fuks-Janczarek, R. Miedziński, E. Gondek, P. Szlachcic and I. V. Kityk, J. Mater. Sci.: Mater. Electron., 2008, 19, 434–441 CrossRef CAS.
- I. Fuks-Janczarek, I. V. Kityk, R. Miedziński, E. Gondek, J. Ebothe, L. Nzoghe-Mendome and A. Danel, J. Mater. Sci.: Mater. Electron., 2007, 18, 519–526 CrossRef CAS.
- M. J. Cho, D. H. Choi, P. A. Sullivan, A. J. P. Akelaitis and L. R. Dalton, Prog. Polym. Sci., 2008, 33, 1013–1058 CrossRef CAS PubMed.
- L. Dalton and S. Benight, Polymers, 2011, 3, 1325–1351 CrossRef CAS PubMed.
- J. Wu, S. Bo, J. Liu, T. Zhou, H. Xiao, L. Qiu, Z. Zhen and X. Liu, Chem. Commun., 2012, 48, 9637 RSC.
- M. He, T. M. Leslie and J. A. Sinicropi, Chem. Mater., 2002, 14, 4662–4668 CrossRef CAS.
- J. Hao, M.-J. Han, K. Guo, Y. Zhao, L. Qiu, Y. Shen and X. Meng, Mater. Lett., 2008, 62, 973–976 CrossRef CAS PubMed.
- J.-M. Raimundo, P. Blanchard, P. Frère, N. Mercier, I. Ledoux-Rak, R. Hierle and J. Roncali, Tetrahedron Lett., 2001, 42, 1507–1510 CrossRef CAS.
- Y. Liang, Y. Wu, D. Feng, S.-T. Tsai, H.-J. Son, G. Li and L. Yu, J. Am. Chem. Soc., 2009, 131, 56–57 CrossRef CAS PubMed.
- J. Hou, M.-H. Park, S. Zhang, Y. Yao, L.-M. Chen, J.-H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- Q. Peng, X. Liu, D. Su, G. Fu, J. Xu and L. Dai, Adv. Mater., 2011, 23, 4554–4558 CrossRef CAS PubMed.
- P. Si, J. Liu, Z. Zhen, X. Liu, G. Lakshminarayana and I. V. Kityk, Tetrahedron Lett., 2012, 53, 3393–3396 CrossRef CAS PubMed.
- Y. V. Pereverzev, K. N. Gunnerson, O. V. Prezhdo, P. A. Sullivan, Y. Liao, B. C. Olbricht, A. J. P. Akelaitis, A. K. Y. Jen and L. R. Dalton, J. Phys. Chem. C, 2008, 112, 4355–4363 CAS.
- D. M. Burland, R. D. Miller and C. A. Walsh, Chem. Rev., 1994, 94, 31–75 CrossRef CAS.
- T. Zhou, J. Liu, G. Deng, J. Wu, S. Bo, L. Qiu, X. Liu and Z. Zhen, Mater. Lett., 2013, 97, 117–120 CrossRef CAS PubMed.
- W. Wu, S. Xin, Z. Xu, C. Ye, J. Qin and Z. Li, Polym. Chem., 2013, 4, 3196–3203 RSC.
- J. Luo, H. Ma, M. Haller, A. K. Y. Jen and R. R. Barto, Chem. Commun., 2002, 888–889 RSC.
- G. Deng, H. Huang, C. Peng, A. Zhang, M. Zhang, S. Bo, X. Liu, Z. Zhen and L. Qiu, RSC Adv., 2014, 4, 4395–4402 RSC.
- W. Wu, Z. Zhu, G. Qiu, C. Ye, J. Qin and Z. Li, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5124–5133 CrossRef CAS.
- W. Wu, C. Li, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, Chem. – Eur. J., 2012, 18, 11019–11028 CrossRef CAS PubMed.
- W. Wu, L. Huang, L. Xiao, Q. Huang, R. Tang, C. Ye, J. Qin and Z. Li, RSC Adv., 2012, 2, 6520–6526 RSC.
- Z. a. Li, W. Wu, C. Ye, J. Qin and Z. Li, Polym. Chem., 2010, 1, 78–81 RSC.
- Z. a. Li, W. Wu, Q. Li, G. Yu, L. Xiao, Y. Liu, C. Ye, J. Qin and Z. Li, Angew. Chem., Int. Ed., 2010, 49, 2763–2767 CrossRef CAS PubMed.
- Q. Zeng, Z. a. Li, Z. Li, C. Ye, J. Qin and B. Z. Tang, Macromolecules, 2007, 40, 5634–5637 CrossRef CAS.
- P. A. Sullivan, H. Rommel, Y. Liao, B. C. Olbricht, A. J. P. Akelaitis, K. A. Firestone, J.-W. Kang, J. Luo, J. A. Davies, D. H. Choi, B. E. Eichinger, P. J. Reid, A. Chen, A. K. Y. Jen, B. H. Robinson and L. R. Dalton, J. Am. Chem. Soc., 2007, 129, 7523–7530 CrossRef CAS PubMed.
- H. Ma, B. Chen, T. Sassa, L. R. Dalton and A. K. Y. Jen, J. Am. Chem. Soc., 2001, 123, 986–987 CrossRef CAS.
- Z. a. Li, Z. Li, C. a. Di, Z. Zhu, Q. Li, Q. Zeng, K. Zhang, Y. Liu, C. Ye and J. Qin, Macromolecules, 2006, 39, 6951–6961 CrossRef CAS.
- H. L. Rommel and B. H. Robinson, J. Phys. Chem. C, 2007, 111, 18765–18777 CAS.
- J. Wu, J. Liu, T. Zhou, S. Bo, L. Qiu, Z. Zhen and X. Liu, RSC Adv., 2012, 2, 1416 RSC.
- D. W. Slocum and P. L. Gierer, J. Org. Chem., 1976, 41, 3668–3673 CrossRef CAS.
- P. Beimling and G. Koßmehl, Chem. Ber., 1986, 119, 3198–3203 CrossRef CAS.
- M. Q. He, T. M. Leslie and J. A. Sinicropi, Chem. Mater., 2002, 14, 2393–2400 CrossRef CAS.
- R. M. Dickson and A. D. Becke, J. Chem. Phys., 1993, 99, 3898–3905 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin and J. C. Burant, Gaussian 03 (Revision B.03), Gaussian Inc., Pittsburgh, PA, 2003 Search PubMed.
- C. Lee and R. G. Parr, Phys. Rev. A, 1990, 42, 193–200 CrossRef CAS.
- K. S. Thanthiriwatte and K. M. Nalin de Silva, J. Mol. Struct.: THEOCHEM, 2002, 617, 169–175 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- C. C. Teng and H. T. Man, Appl. Phys. Lett., 1990, 56, 1734 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and HRMS spectra of resulted compounds. See DOI: 10.1039/c4ra01767f |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 




