Borontribromide-mediated C–C bond formation in cyclic ketones: a transition metal free approach†
Imran Ahmada,
Vinay Pathaka,
Prema G. Vasudevb,
Hardesh K. Mauryaa and
Atul Gupta*a
aMedicinal Chemistry Department, Central Institute of Medicinal and Aromatic Plants, PO. CIMAP, Kukrail Road, Lucknow-226015, India. E-mail: atisky2001@yahoo.co.in; Tel: +91 5222718556
bMetabolic & Structural Biology Department, CSIR-Central Institute of Medicinal and Aromatic Plants, P.O. CIMAP, Kukrail Road, Lucknow-226015, India
First published on 13th May 2014
Abstract
Borontribromide (BBr3) is a well-known demethylating agent. The current investigation was focused on a new application of borontribromide as a C–C bond forming agent in cyclic ketones. In this study, borontribromide mediated C–C bond formation reactions of tetralones, chromenone, thiochromenone and indanones were explored. A methoxy group containing ketones showed selective C–C bond formation reaction instead of demethylation of the methoxy group. MM2 steric energy calculations for the final products showed that the reaction favored the formation of exo- or endo-cyclic double bond containing products, depending upon their low MM2 steric energy in a specific frame structure, as observed in X-ray crystallography. A comprehensive crystallographic and pi-stacking analysis of product 10a demonstrated the formation of 10a as an enantiomeric mixture, and its centre of inversion was stabilized by a set of three unique pi–pi interactions.
Introduction
Expensive and rarely available transition metal catalyzed C–C bond forming reactions are an attractive research topic at present.1 Due to the expense and limited availability of transition metals, there is a need to search for alternate methods for constructing C–C and other bonds.2 Borontribromide is a commercially available Lewis acid and a well-known demethylating or dealkylating agent used for the cleavage of ethers, subsequent cyclization and often in the production of pharmaceuticals.3 It is also used as a catalyst in Friedel–Craft reactions, olefin polymerization and in the manufacture of semiconductors as a boron source.4 During the synthesis of important type II tetralone-based antiestrogenic molecules (Fig. 1), we observed BBr3 mediated C–C bond formation instead of demethylation. We herein report this new application of borontribromide (BBr3) as a C–C bond forming reagent, which has not been reported previously. | ||
| Fig. 1 Targeted estrogenic/anti-estrogenic molecules. | ||
Results and discussion
Chemistry
Our primary strategy to synthesize target molecules (II, Fig. 1) similar to the estradiol derivative (I)5 was based on the conventional approach for the total synthesis of steroidal estrogens starting from tetralones, i.e. Torgov synthesis. For this approach, we planned to synthesize substituted 3-(4-hydroxyphenyl)tetralones as intermediates.The synthesis of 3-(4-hydroxyphenyl)tetralone started with a Friedel–Craft acylation reaction on anisole (1) using phenylacetic acid (2) in polyphosphoric acid (PPA), which yielded 4-methoxy deoxybenzoin (3) in good yield.6 Subsequently, a Reformatsky reaction on compound 3 with ethyl bromoacetate and zinc dust in dry diethyl ether and benzene mixture yielded compounds 4 and 5 (Scheme 1).7
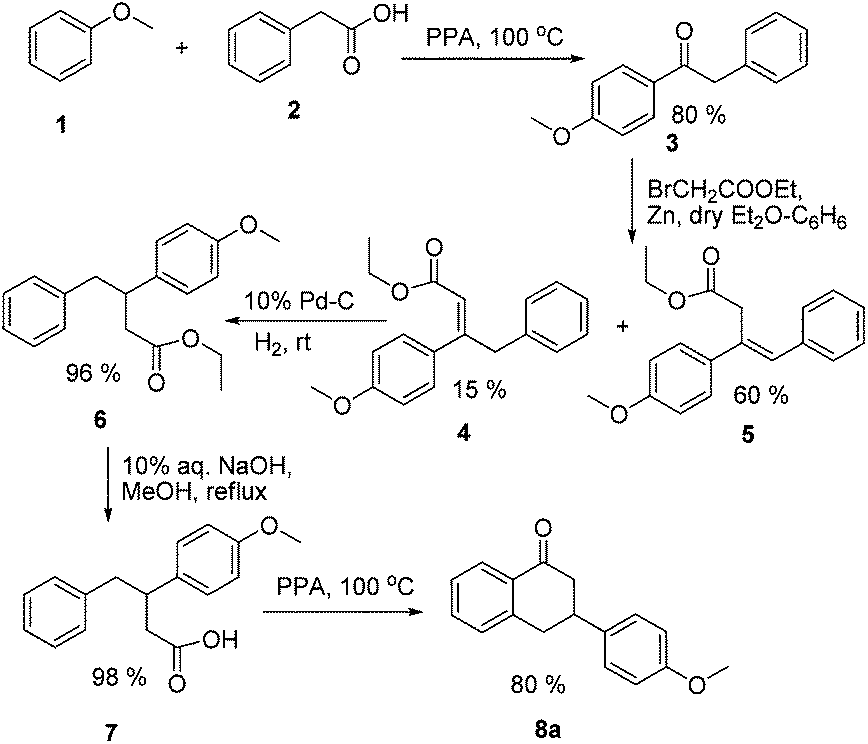 | ||
| Scheme 1 Synthesis of precursor (8a). | ||
Both compounds 4 and 5 were subjected to catalytic hydrogenation using 10% palladium on charcoal (Pd–C) and hydrogen gas at room temperature, which gave a common reaction product 6. Note that due to the approximately same Rf of 5 and 6, the reaction was monitored by 1H NMR.8 Alkaline hydrolysis of compound 6 yielded a corresponding acid derivative (7) in quantitative yield. Thereafter, compound 7 upon intramolecular cyclization in the presence of PPA yielded 3-(4-methoxyphenyl)tetralone (8a) in 80% yield (Scheme 1).
In our initial attempts to transform 3-(4-methoxyphenyl)tetralone (8a) into the corresponding hydroxy derivative (9), we used pyridinium hydrochloride as a demethylating agent over a temperature range of 160–170 °C, which gave the desired compound (9) in 85% yield (Table 1). Due to the high temperature of this reaction, we explored other demethylating agents that were reported in the literature, which could be used under normal reaction conditions. In our attempts, we used borontribromide (BBr3), which is another versatile demethylating agent used with a variety of substrates at low temperature.
| S.no. | Catalyst/solvent | Temp. (°C) | Time (min) | Yield of 9 (%) | Yield of 10a (%) |
|---|---|---|---|---|---|
| a No solvent.b DCM under N2.c DCM without N2.d Product is not formed.e 3 equivalent.f 2 equivalent.g 1 equivalent.h No reaction. | |||||
| 1 | C5H5N·HCla | 160–170 | 60 | 85 | d |
| 2 | BBr3b,e | −78 | 70 | d | 81 |
| 3 | BBr3c,e | −78 | 70 | d | 80 |
| 4 | BBr3b,e | −30 | 110 | d | 80 |
| 5 | BBr3c,e | −30 | 110 | d | 81 |
| 6 | BBr3c,e | −20 | 150 | d | 82 |
| 7 | BBr3c,e | 0 to rt | 180 | d | 82 |
| 8 | BBr3c,f | 0 to rt | 24 h | d | 53 |
| 9 | BBr3c,g | 0 to rt | 24 h | d | 29 |
| 10 | BF3OEtb | 0 to rt | 24 h | h | h |
| 11 | BF3OEtc | 0 to rt | 24 h | h | h |
| 12 | AlCl3c | 0 to rt | 24 h | h | h |
Interestingly, under these reaction conditions, the attempted demethylation using BBr3 yielded endocyclic β–γ unsaturated diastereomeric partially reduced bisnaphthalene (10a)9 instead of demethylated product 9 (Scheme 2, Table 1).
 | ||
| Scheme 2 Synthesis of product 9 and 10a. | ||
The formation of compound 10a was optimized at different temperatures with BBr3 and other Lewis acids (Table 1).
In order to optimize the reaction conditions, we attempted the reaction with different molar equivalents of BBr3 in DCM from 0 °C to room temperature. It was observed that the use of one equivalent of BBr3 yielded only 10a in 37% yield along with unreacted starting material (8a) even at an extended reaction time (24 h, Table 1). Similarly, the use of two equivalents of BBr3 gave only the compound 10a in 42% yield with recovery of 8a in 24 h. These results showed that the use of three equivalents of BBr3 was required to complete the reaction with a better yield of product 10a (Table 1). Furthermore, the suitability of the solvent for the reaction was examined using different solvents such as benzene, toluene, tetrahydrofuran (THF), dioxane and ethanol. Of these solvents, the reaction in toluene could only produce product 10a in 70% yield in 5 h, whereas the reaction could not proceed in other solvents (Table 2). Therefore, it was concluded that use of three equivalents of BBr3 in DCM from 0 °C to room temperature for the observed C–C bond forming reaction was the optimal reaction condition.
| S.no. | Catalyst | Solvent | Temp. (°C) | Time (h) | Yield of 10c (%) |
|---|---|---|---|---|---|
| a Not reported.b Using Dean–Stark trap.c 3 equivalent.d No reaction. | |||||
| 1 | (t-BuO)3Al | Toluene | Reflux | 12 | 8 (ref. 11) |
| 2 | Amberlyst-15 | Xylene | Reflux | a | 20 (ref. 12) |
| 3 | TiCl4, Et3N | Hexane, DCM, N2 | −10 | a | 70 (ref. 12) |
| 4 | TiCl4 | DCE | Reflux | a | 71 (ref. 13) |
| 5 | Amberlyst-15 | Toluene | Refluxb | 4.5 | 75 (ref. 14) |
| 6 | BBr3c | DCM | 0 to rt | 3 | 80 |
| 7 | BBr3c | THF | 0 to rt | 24 | d |
| 8 | BBr3c | 1,4-Dioxane | 0 to rt | 24 | d |
| 9 | BBr3c | Benzene | 0 to rt | 24 | d |
| 10 | BBr3c | Toluene | 0 to rt | 5 | 70 |
| 11 | BBr3c | Ethanol | 0 to rt | 24 | d |
An extensive literature survey revealed that various reagents, such as Al2O3,10 (t-BuO)3Al,11 HCl, amberlyst-29, CH3SO3H, NaOH, Triton-B, p-dimethylaminopyridine,12 TiCl4,12,13 and amberlyst-15,12,14 have been explored for C–C bond formation of 1-tetralone (8c). It was found that transition metal containing reagents were primarily applied to explore such reactions.12,13,15,16
Considering this, we were interested to explore BBr3-mediated methodology for C–C bond forming reactions in cyclic ketones at low temperature, which could be superior to other reported methods.
To establish the generality of this method, we explored various reactions of substituted tetralones and other six member ketones such as chromanone and thiochromanone as reported in Scheme 3 (Table 3).
 | ||
| Scheme 3 Synthesis of BBr3-mediated partially reduced bisnaphthalenes (10a–f), bischromane 10g and bisthiochromane 10h. | ||
| Comp. | Product | Reaction time | Mp (°C) | Yield |
|---|---|---|---|---|
| 10a | 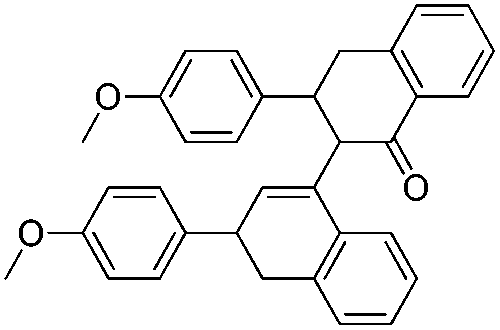 |
3 h | 192–193 | 82 |
| 10b |  |
8 h | 182–183 | 75 |
| 10c |  |
4 h | 131–132 | 80 |
| 10d |  |
4 h | 117–118 | 83 |
| 10e |  |
8 h | 130–131 | 73 |
| 10f |  |
3 h | 182–183 | 72 |
| 10g |  |
9 h | 127–128 | 70 |
| 10h |  |
8 h | 119–120 | 73 |
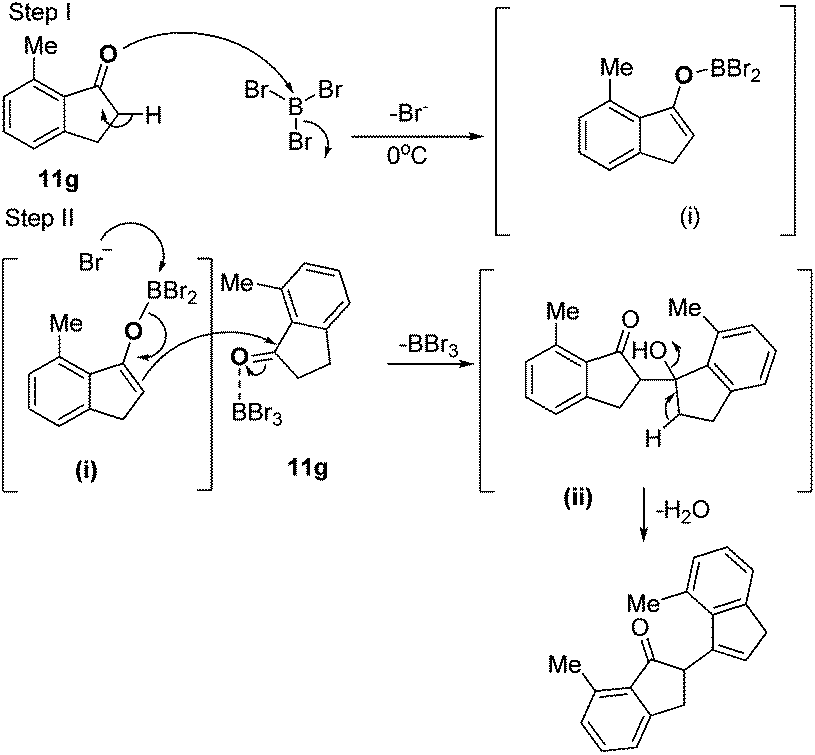
The plausible mechanism reveals that the reaction starts by activation of the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group of cyclic ketone 8a by BBr3, followed by formation of an intermediate (i, step I). The enolic intermediate (i) attacks the other molecule of starting precursor 8a (it was assumed that the molecule 8a was pre-activated by excess BBr3 present in the reaction) with elimination of BBr3 and subsequently eliminates H2O from intermediate ii, leading to the formation of product 10a (step II) as depicted in Scheme 4.
O) group of cyclic ketone 8a by BBr3, followed by formation of an intermediate (i, step I). The enolic intermediate (i) attacks the other molecule of starting precursor 8a (it was assumed that the molecule 8a was pre-activated by excess BBr3 present in the reaction) with elimination of BBr3 and subsequently eliminates H2O from intermediate ii, leading to the formation of product 10a (step II) as depicted in Scheme 4.
 | ||
| Scheme 4 Plausible mechanism for the formation of 10a. | ||
Furthermore, we extended the applicability of BBr3 as a C–C bond forming agent to a cyclic constrained ketone molecule with structural similarities with 10a. Thus, 3-(4-methoxyphenyl)indanone (11a) was selected, and upon reaction with BBr3 under similar reaction conditions it yielded 12a with an exocyclic α–β unsaturated ketone system instead of demethylated product 13 (Scheme 5).
 | ||
| Scheme 5 Synthesis of bisindene (12a) and selective demethylated product (16): (a) BBr3, dry dichloromethane, −78 °C to room temp, 5 h, (b) (CH3)2SO4, anhydrous K2CO3, dry acetone, 12 h. | ||
To elucidate the impact of steric factor at C-2 of the ketones, we further explored the reaction of BBr3 with an indanone analog (15), which was synthesized from precursor 14 and had a bulky phenyl group at the α-position (Scheme 5). Instead of a C–C bond containing product, this reaction resulted in the formation of monodemethylated product (16) selectively. Thus, the α-CH2 group was necessary for the C–C bond formation reaction and in the absence of an α-CH2 group, the reaction proceeded towards selective demethylation. Possibly, the presence of a phenyl group at the C-2 position in 15 created steric hindrance and did not pave the way for another molecule to yield bisindene. Consequently, the reaction was forced to proceed toward demethylation rather than C–C bond formation (Scheme 5). It is worthwhile to mention that demethylation of 3-(4-methoxyphenyl)indanone analogs catalyzed with BBr3 have not been reported in the literature. However, BBr3-mediated demethylation of 2-aryl substituted 3-(4-methoxyphenyl)indanone analogs have been reported in the literature.17 In general, various reagents such as HBr–AcOH, LiBr–DMF, and Py·HCl have been the preferred choice for the demethylation of indanone derivatives.18
On the basis of the results presented above (12a, Scheme 5), we have further applied our methodology to different substituted indanones (11a–g), which yielded compounds 12a–f and 17 in good yields (Schemes 6 and 7, Table 4).
 | ||
| Scheme 6 BBr3-catalyzed synthesis of partially reduced bisindene (12a–f) with a plausible mechanism. | ||
 | ||
| Scheme 7 BBr3-mediated synthesis of partially reduced endocyclic double bond containing bisindene (17) with a plausible mechanism. | ||
| Comp. | Product | Reaction time | Mp (°C) | Yield |
|---|---|---|---|---|
| 12a |  |
3 h | 62–63 | 80 |
| 12b |  |
4 h | 212–213 | 74 |
| 12c |  |
3 h | 175–176 | 89 |
| 12d |  |
4 h | 157–158 | 72 |
| 12e |  |
1 h | 231–232 | 91 |
| 12f |  |
2 h | 164–165 | 90 |
| 17 |  |
2 h | 112–113 | 86 |
Compounds 12a–f were formed due to selective deprotonation from the α-CH2 of ii as shown in Scheme 6, whereas 17 was formed due to selective deprotonation from the γ-CH2 of ii as shown in Scheme 7.
Interestingly, it was observed that in the case of indanones (11a–f), the position of the double bond was exocyclic with an α–β unsaturated ketone system, whereas with the six membered cyclic ketones (8a–h), the position of the double bond was endocyclic with a β–γ unsaturated ketone system exclusively.
However, 2,3-dihydro-7-methylinden-1-one (11g) yielded endocyclic β–γ unsaturated partially reduced bisindene (17, Scheme 7, Fig. 2) as observed in the case of six membered ketones. Such an anomaly with indanone (17) could have been due to possible intramolecular steric interactions between CO and the 7-substituted methyl group, which was less in a β–γ unsaturated arrangement than an α–β unsaturated system.
 | ||
| Fig. 2 (a) Molecular conformation in the crystal of 10a with 50% thermal ellipsoids; (b) view down the C1′–C2 bond of 10a, indicating the orientation of the two halves within 10a and the aromatic rings shown as I–IV; (c) molecular conformation in the crystal of 12f with 50% thermal ellipsoids; and (d) molecular conformation in the crystal of 17 with 50% thermal ellipsoids. | ||
In general, the ring size of ketones had a great impact on the position of CC bonds in products. However, in the case of compound (17), the position of the substituent had a more pronounced effect on the position of the CC bond than the ring size of the ketone.
On the basis of the results presented above, we further explored the cross reactions between tetralones and indanones under these reaction conditions (Scheme 8). The cross reaction of tetralones 8d and 8f under these reaction conditions gave self-condensed products 10d and 10f along with inseparable cross products 18a and b in 25%, 26%, and 27% yields, respectively. Similarly, cross reaction between indanones 11c and 11b yielded self-condensed products 12c and 12b along with inseparable cross products 19a and b in 30%, 29% and 31% yields, respectively. However, cross reaction between tetralone (8d) and indanone (11c) gave only self-condensed products 10d and 12c in 37% and 33% yields, respectively, which could be presumably due to the stereochemistry of tetralone and indanone molecules and their selectivity for the formation of exo- versus endo-cyclic double bond containing products (discussed below in detail).
 | ||
| Scheme 8 BBr3-mediated cross reactions between different cyclic ketones. | ||
As observed above in the case of tetralone (8a–h) and indanone (11g), which have the same tendency to yield endocyclic double bond containing products despite their differences in stereochemistry, indanone 11g and tetralone 8d were selected for their cross reactivity. The reaction between tetralone (8d) and indanone (11g) yielded self-condensed products 10d and 17 along with crossed products 20 and 21 in 18%, 27%, 23% and 20% yields, respectively. The formation of cross products 20 and 21 could be due to the similar reactivity of tetralone 8d and indanone 11g to yield endocyclic double bond containing products.
However, the attempted C–C bond forming reaction in substituted acetophenones using BBr3 in DCM at 0 °C to room temperature did not yield any product, but rather unreacted starting material was recovered. Interestingly, a methoxy group containing acetophenone yielded only demethylated product.
Possible explanation for the formation of a C–C bond in 8a instead of demethylation
In compound 8a, the donation of a lone pair of electrons by two O-atoms, one from a CO group and another from a methoxy group, was possible (Scheme 2). Due to the bulky nature of the phenyl group substituted at C-3 (either in equatorial or axial orientation), shifting of a lone pair of electrons from the O-atom of CO would have been easier than from the O-atom of the methoxy group, resulting in the formation of intermediate i (Schemes 4, 6 and 7). This phenomenon would have forced the reaction for C–C bond formation instead of demethylation. Furthermore, in both situations (axial or equatorial), compound 8a had enough space around the C-2 atom to accommodate another 8a molecule to form partially reduced bisnaphthalenes (10a).
In the case of 12a, the pentanone ring fused with benzene had a distorted envelop shape wherein a carbonyl carbon remained on the top of the envelop and a phenyl ring attached to C-3. In this case also, compound 11a had enough space around C-2 to accommodate another molecule of 11a to yield bisindene (12a).
The selective demethylation of compound 15 by BBr3 under similar reaction conditions could be assumed due to the presence of a bulky phenyl group at C-2, which could have hampered the adjustment of another molecule of indanone at the C-2 position.
Explanation of exo- versus endo-cyclic double bond formation through computational MM2 steric energy study
To get an approximation for the formation of exo- versus endo-cyclic double bond containing products, the computational MM2 steric energy calculations for 10, 12 and 17 were made using chemBio3D ultra 11.0 (ESI Table S1†). The representative frame structures of 10, 12 and 17 (ESI Table S1†) were drawn on the basis of the model frame X-ray crystal structure of 10a, 12f and 17 (as a standard, Fig. 2). The results showed that (a) six membered cyclic ketones (8a–h) favored the formation of endocyclic double bond containing products (10a–h) stabilized with low steric energy, (b) five membered cyclic ketones (11a–f) favored the formation of exocyclic double bond containing products (12a–f) stabilized with low steric energy and (c) five membered cyclic ketone (11g) favored the formation of an endocyclic double bond containing product (17) stabilized with low steric energy.Thus, MM2 steric energy calculations of products showed that the reaction favored the formation of exo- or endo-cyclic double bond containing products, depending upon their low MM2 steric energy for a specific frame structure as obtained from X-ray crystallography.
X-ray diffraction studies of 10a and 17
For X-ray analysis, crystals of 10a were obtained by dissolving it in dichloromethane and the subsequent slow evaporation of solvent. Similarly, crystals of 12f were obtained by slow evaporation from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of chloroform and hexane, whereas crystals of compound 17 were obtained by slow evaporation of solvent (EtOAc–hexane; 1:25) at room temperature (15–18 °C).19 The molecular structures of the compound 10a, 12f and 17 with arbitrary numbering are represented in Fig. 2.
:1 mixture of chloroform and hexane, whereas crystals of compound 17 were obtained by slow evaporation of solvent (EtOAc–hexane; 1:25) at room temperature (15–18 °C).19 The molecular structures of the compound 10a, 12f and 17 with arbitrary numbering are represented in Fig. 2.
The crystal structure of 10a shows a non-planar, distorted conformation of the molecule. The crystal structure determination revealed that compound 10a had a centrosymmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , in which the two enantiomers were related by an inversion centre.
, in which the two enantiomers were related by an inversion centre.
The molecular conformation of 10a in the asymmetric unit is shown in Fig. 2a. The relative configuration of the newly generated chiral centre at C2 was found to be same as C3 (and C3′) of the product. A view of the molecule down the C1′–C2 bond clearly demonstrated the orientation of the two halves of the molecule and the relative disposition of the aromatic rings as shown in Fig. 2b. The 3-(4-methoxyphenyl)tetralone moieties were oriented at about 70° with respect to each other. Furthermore, 4-methoxyphenyl rings of 10a were oriented perpendicular to the corresponding tetralone rings. Moreover, although 10a had four aromatic rings, there were no strong intramolecular interactions involving them.
Pi-stacking study of compound 10a
Pi-stacking refers to attractive, noncovalent interactions between aromatic rings and is also called as π–π stacking.20 It is applied in materials science21 as the molecular receptor of a guest molecule in supramolecular chemistry22 and various other applications. Because compound 10a had four aromatic rings, it provided an opportunity to investigate aromatic interactions within the molecule and between molecules in the supramolecular assembly.The aromatic rings in the molecule were labelled I–IV as shown in Fig. 2b. The relevant parameters for intra- and inter-molecular π–π interactions stabilizing the crystal structure are shown in Table 5.
| Type | Cg⋯Cg (Å) | C⋯C shortest (Å) | Interplanar angle (°) | Symmetry |
|---|---|---|---|---|
| a Aromatic ring labels (I–IV) as shown in Fig. 2b. Cg = centre of gravity. Intermolecular π⋯π interactions are labelled (A)–(J) for clarity in discussion. | ||||
| Intramolecular | ||||
| III–IV | 5.53 | 4.19 (C15–C16′) | 67.2 | |
| II–III | 5.77 | 3.78 (C11–C10′) | 47.9 | |
| Intermolecular | ||||
| I–II (A) | 5.31 | 3.93 (C5–C5′) | 70.0 | −x + 1, −y + 1, −z + 1 |
| I–III (B) | 4.84 | 3.66 (C7–C13) | 85.8 | −x + 1, −y + 1, −z + 1 |
| II–III (C) | 5.22 | 3.72 (C8′–C16) | 48.2 | −x + 1, −y + 1, −z + 1 |
| I–I (D) | 5.37 | 3.83 (C9–C9) | 0.2 | −x + 2, −y + 1, −z + 1 |
| I–III (E) | 5.86 | 3.82 (C8–C12) | 85.8 | −x + 2, −y + 1, −z + 1 |
| III–IV (F) | 5.53 | 3.66 (C15–C14′) | 67.2 | −x + 2, −y + 1, −z |
| II–II (G) | 4.03 | 3.32 (C7′–C7′) | 1.3 | −x + 1, −y + 2, −z + 1 |
| IV–IV (H) | 5.23 | 3.43 (C16′–C16′) | 0.0 | −x + 2, −y + 2, −z |
| IV–II (I) | 5.64 | 3.71 (C14′–C5′) | 73.6 | −x + 1, −y + 2, −z |
| IV–I (J) | 5.79 | 3.58 (C6–C13′) | 26.4 | x, y + 1, z − 1 |
The enantiomeric pair of molecules generated around the centre of inversion was stabilized by a set of three unique π–π interactions between groups I and II, I and III, and II and III (Fig. 3).
 | ||
| Fig. 3 Enantiomeric pair of 10a stabilized by aromatic π–π interactions (pi-stacking). Interactions are labelled as described in Table 5. | ||
The interaction I–III had an edge-to-face geometry with the ring planes inclined at an angle of 85°. However, no C–H–π interaction was present between the two rings (C–H–Cg = 2.8 Å, ∠C–H–Cg = 128°). The centrosymmetric products, thus formed, were connected through weak π–π, C–H–O and C–H–π interactions, resulting in the observed three-dimensional assemblies in crystals (Fig. 4 and 5).
 | ||
| Fig. 4 A view of the molecular packing of 10a down the crystallographic b-axis showing π–π (black-dotted lines) and C–H–π (blue-dotted lines) stacking. | ||
 | ||
| Fig. 5 C–H–O interactions of 10a observed in the crystal. | ||
Weak π–π interactions, listed in Table 5, had centre–centre distances ranging from 4.0–5.9 Å, and the shortest C⋯C distances range from 3.32–3.93 Å. Based on the distances, it could be concluded that the interaction between tetralone rings II according to the symmetry −x + 1, −y + 2, −z + 1 (G in Table 5) was the strongest π–π interaction in the crystal. Although interactions B and E had a T-shaped geometry (edge-to-face), no C–H–π interactions were observed in any case. Interactions D, G and H had parallel displaced orientation of the aromatic panes, while the other interactions had an inclined geometry (Table 5).
There were two C–H–O hydrogen bonds, one between the carbonyl oxygen and tetralone ring II [C6′–H–O1 (−x + 1, −y + 2, −z + 1); C6′–O1 = 3.41 Å, H–O1 = 2.54 Å, ∠C6′–H–O1 = 150.7°; Fig. 5] and another between the tetralone ring I and methoxy oxygen atom O2′ [C8–H–O2′ (x, y, z + 1); C8–O2′ = 3.40 Å, H–O2′ = 2.49 Å, ∠C8–H–O2′ = 158°; Fig. 5], which further stabilized the packing of molecules along the crystallographic b and c axes. A weak C–H–π interaction was also observed between the C2–H and tetralone ring I of molecules related by the symmetry −x + 2, −y + 1, −z + 1 (C2–Cg = 3.734 Å, H–Cg = 2.84 Å, C–H–Cg = 147°; Fig. 4).
In order to gain insight into the entrapping behavior of 10a toward a host–guest relationship, we studied space filling diagrams of 8a as well as 10a (Fig. 6). The pendent methoxyphenyl groups in 10a were inclined to each other (approximately 70°), leaving little space between them for solvent molecules (or other small molecules) to be entrapped. Similarly, a close-packing of the centrosymmetrically related molecules was observed, which was stabilized by multiple aromatic interactions in 10a.
 | ||
| Fig. 6 Space-filling diagram (A) and centrosymmetric diagram (B) of compound 10a. Two inversion-related molecules are shown in different colours. | ||
Furthermore, compound 17 was crystallized as a racemic mixture in the centrosymmetric space group P. The two halves of the molecule were oriented perpendicular to each other with a dihedral angle about the C2–C11 bond being 85°. The bond length of C11–C12 was 1.347 Å, and the bond angle C2–C11–C12 was 124°, which confirmed the double bond character of C11–C12.
The molecular packing in the crystal of 17 was stabilized by aromatic π–π interactions between inversion related molecules and a weak C–H–O interaction between the translated molecules along the crystallographic a-axis (Fig. 7; ESI Table S2†).
 | ||
| Fig. 7 Two views of the molecular packing in the crystal of 17. Aromatic interactions (top) and hydrogen bonds (bottom) are indicated as dotted lines. Distances between the centroids of interacting aromatic rings are also indicated. The C–H–O hydrogen bond has the following parameters: H–O = 2.42 Å, ∠C–H–O = 146.6°. | ||
Conclusion
In conclusion, the current investigation disclosed a new application of BBr3 as a C–C bond forming agent in cyclic ketones such as tetralone, chromanone, thiochromenone and indanone derivatives. A detailed study revealed that α-methylene containing cyclic ketone (with a methoxy group) preferred C–C bond formation instead of demethylation, and in the absence of the α-methylene group, the reaction proceeded towards selective demethylation. MM2 steric energy calculations of the products showed that the reaction favored the formation of exo- or endo-cyclic double bond containing products, depending upon their low MM2 steric energy for a given frame structure.Overall, an efficient and convenient borontribromide (BBr3) mediated methodology for C–C bond formation reactions of cyclic ketones has been developed. The stereoselective C–C bond formation using borontribromide (BBr3) is under study and will be reported in the future.
Experimental section
General
The reagents and solvents used in this study were of analytical or laboratory grade and used without further purification unless stated otherwise. All the reactions were monitored on Merck aluminium silica gel thin layer chromatography (TLC, UV254nm) plates. Column chromatography was carried out on silica gel (100–200 or 230–400 mesh). The melting points were determined on a Buchi melting point M560 apparatus in open capillaries and are uncorrected. Commercial reagents were used without purification. 1H and 13C NMR spectra were recorded on a Bruker WM-300 (300 MHz) using CDCl3, acetone-D6 or DMSO-d6 as the solvent. Chemical shifts are reported in parts per million (ppm) shift (δ-value) based on the middle peak of the solvent (CDCl3, acetone-D6 and DMSO-d6). Signal patterns are indicated as s, singlet; bs, broad singlet; d, doublet; dd, double doublet; t, triplet; and m, multiplet. Coupling constants (J) are given in Hertz. Infrared (IR) spectra were recorded on a Perkin-Elmer AX-1 spectrophotometer in a KBr disc and reported in wave number (cm−1). ESI-MS mass spectra were recorded on a Shimadzu LC-MS and/or LC-MS-MS APC3000 (Applied Biosystems).General procedure for the synthesis of (3)
Compounds 1 (20 g, 146.9 mmol) and 2 (15.99 mL, 146.9 mmol) were placed in a round-bottomed flask, and polyphosphoric acid (PPA, 140.0 g) was added to it. The reaction mixture was heated at 100 °C on a water bath. The progress of the reaction was monitored using TLC (30% ethyl acetate–hexane solvent system). After completion of the reaction, the reaction mixture was poured on ice and extracted by ethyl acetate. The organic layer was washed with sodium bicarbonate (NaHCO3) solution. The organic layer was then separated, dried over anhydrous sodium sulphate (Na2SO4) and concentrated. The crude material was purified by column chromatography using silica gel (100–200 mesh size) and hexane–ethyl acetate (49:1), which yielded pure compound 3 as a white solid in 80% yield; Rf: 0.60 (3:7 ethyl acetate–hexane); mp: 74–75 °C; IR (KBr, νmax/cm−1): 1680 (CO), 160; 1H NMR (CDCl3, 300 MHz, δ ppm): 3.86 (s, 3H, OCH3), 4.45 (S, 2H, CH2), 6.94 (d, J = 9.0 Hz, 2H, ArH), 7.15 (m, 5H, ArH), 8.28 (d, J = 8.7 Hz, 2H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 45.68 (CH2), 55.88 (OCH3); 114.21 (2xC), 127.19, 129.05 (2xC), 129.80 (2xC), 130.05 (2xC), 131.37 (2xC), 135.39, 163.94, 196.66 (CO); m/z: 249 [M + Na]+.
General procedure for the synthesis of ethyl-3-(4-methoxyphenyl)-4-phenylbut-2-enoate (4) and ethyl-3-(4-methoxyphenyl)-4-phenylbut-3-enoate (5)
In a three neck round-bottomed flask fitted with a condenser and nitrogen gas supply, zinc dust (Zn, 14.5 g, 221.2 mmol) was activated by heating it in the presence of a few crystals of iodine (I2). Dry diethyl ether was added to the flask followed by dropwise addition of ethyl bromoacetate (25.48 mL, 221.2 mmol) with mild heating. A vigorous reaction occurred, and the reagent was formed. Compound 3 (10 g, 44.24 mmol), which was dissolved in dry benzene, was added to this mixture dropwise at room temperature. Subsequently, the reaction was transferred to oil bath apparatus for refluxing. The progress of the reaction was monitored by TLC (10% ethyl acetate–hexane). After completion of the reaction, the reaction mixture was treated with dilute HCl and worked up using ethyl acetate and water. The organic layer was the separated, dried over anhydrous sodium sulphate (Na2SO4) and concentrated. The crude material was purified by column chromatography using silica gel (100–200 mesh size) and ethyl acetate–hexane (0.5:99.5 for 4 and 1.0:99 for 5) as the eluent, which yielded pure compounds 4 and 5 in 15% and 60% yields, respectively.
O); 297 [MH+].O); 1H NMR (CDCl3, 300 MHz, δ ppm): 1.18 (t, J = 6.9 Hz and 7.2 Hz, 3H, CH3), 3.69 (s, 2H, CH2), 3.89 (s, 3H, OCH3), 4.12 (q, 2H, CH2), 6.92 (d, J = 9.0 Hz, 2H, ArH), 6.97 (s, 1H, CH), 7.26 (m, 1H, ArH), 7.37 (m, 4H, ArH), 7.46 (dd, J = 2.1 Hz and 1.5 Hz 2H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 14.52 (CH3), 37.07 (CH2), 55.71 (OCH3), 61.18 (CH2), 114.25 (2xC), 127.41, 127.76 (2xC), 128.80 (2xC), 129.16 (2xC), 130.13, 134.48, 134.53, 138.06, 159.62, 172.06 (CO); 297 [MH+].General procedure for the synthesis of ethyl 3-(4-methoxyphenyl)-4-phenylbutanoate (6)
Compound 4 or 5 (14.0 g, 47.30 mmol) was placed in a round-bottomed flask and dissolved in an ethyl acetate and methanol mixture (7:3). The mixture was flushed with nitrogen gas. Palladium charcoal (Pd/C, 5 wt%) (4.0 g) was added, and hydrogen gas was passed for 24 h. The progress of the reaction was monitored by TLC (10% ethyl acetate–hexane) and 1H NMR. After completion of the reaction, the solvent was evaporated and crude material was worked up using ethyl acetate and water. The organic layer was then separated, dried over anhydrous sodium sulphate (Na2SO4) and concentrated. The crude material was purified by column chromatography using silica gel (100–200 mesh size) and ethyl acetate–hexane (0.5:99.5) as the eluent, which yielded pure compound 6 as oil in 96% yield; Rf: 0.27 (10% ethyl acetate–hexane); IR (KBr, νmax/cm−1): 1733 (CO); 1H NMR (acetone-D6, 300 MHz, δ ppm): 1.08 (t, J = 7.2 Hz and 7.2 Hz, 3H, CH3), 2.59 (m, 2H, CH2), 2.93 (m, 2H, CH2), 3.36 (m, 1H, CH), 3.72 (s, 3H, OCH3), 3.93 (q, 2H, CH2), 6.80 (d, J = 8.7 Hz, 2H, ArH), 7.16 (m, 7H, ArH); 13C NMR (acetone-D6, 75 MHz, δ ppm): 13.98, 40.71 (CH2), 42.97 (CH2), 43.75, 54.88, 59.96 (CH2), 113.88, 126.31, 128.45 (2xC), 129.55, 135.95, 140.54, 158.72 (phenolic), 171.88 (CO); mass: 283 [M − CH3]+, 337 [M + K]+.
General procedure for the synthesis of 3-(4-methoxyphenyl)-4-phenylbutanoaic acid (7)
In a round-bottomed flask, compound 6 (10 g, 33.55 mmol) was dissolved in methanol, and 10% aqueous sodium hydroxide (NaOH) solution (70 mL) was added. The reaction mixture was transferred to a reflux apparatus. The progress of the reaction was monitored by TLC (30% ethyl acetate–hexane). After completion of the reaction, the solvent was evaporated and the reaction mixture was treated with dilute HCl and worked up using ethyl acetate and water. The organic layer was then separated, dried over anhydrous sodium sulphate (Na2SO4) and concentrated. The crude solid material was purified by washing it with hexane, which yielded pure compound 7 as an off-white solid in 98% yield; Rf: 0.36 (30% ethyl acetate–hexane); mp: 124–125 °C; IR (KBr, νmax/cm−1): 3424, 3027, 2933, 1706 (CO), 1609, 1513, 1437, 1242, 1033, 961, 822, 700; 1H NMR (acetone-D6, 300 MHz, δ ppm): 2.59 (m, 2H, CH2), 2.93 (m, 2H, CH2), 3.37 (m, 1H, CH), 3.73 (s, 3H, OCH3), 6.80 (d, J = 8.7 Hz, 2H, ArH), 7.16 (m, 7H, ArH); 13C NMR (acetone-D6, 75 MHz, δ ppm): 40.22 (CH2), 42.99 (CH2), 43.59, 54.86, 113.88, 126.25, 128.41, 128.96 (2xC), 129.54 (2xC), 136.14, 140.62, 158.69 (phenolic), 172.95 (CO); mass: 269 [M − H]+.
General procedure for the synthesis of 3,4-dihydro-3-(4-methoxyphenyl)naphthalene-1(2H)-one or 3-(4-methoxyphenyl)tetralone (8a)
Compound 7 (10 g, 37.04 mmol) was placed in a round-bottomed flask, and polyphosphoric acid (PPA, 70.0 g) was added to it. The reaction mixture was heated at 100 °C on a water bath. The progress of the reaction was monitored using TLC (30% ethyl acetate–hexane solvent system). After completion of the reaction, the reaction mixture was poured on ice and extracted by ethyl acetate. The organic layer was washed with sodium bicarbonate (NaHCO3) solution. The organic layer was then separated, dried over anhydrous sodium sulphate (Na2SO4) and concentrated. The crude material was purified by column chromatography using silica gel (100–200 mesh size) and ethyl acetate–hexane (4:96), which yielded pure compound 8 as an off-white solid in 80% yield; Rf: 0.86 (30% ethyl acetate–hexane); mp: 88–89 °C; IR (KBr, νmax/cm−1): 1679 (CO); 1H NMR (acetone-D6, 300 MHz, δ ppm): 2.81 (m, 2H, CH2), 3.20 (m, 2H, CH2), 3.38 (m, 1H, CH), 3.77 (s, 3H, OCH3), 6.91 (d, J = 8.7 Hz, 2H, ArH), 7.41 (m, 4H, ArH), 7.55 (m, 1H, CH), 7.97 (d, J = 9.0 Hz, 1H, ArH); 13C NMR (acetone-D6, 75 MHz, δ ppm): 37.85 (CH2), 40.63 (2C), 46.30 (CH2), 55.02, 114.33 (2xC), 126.85, 127.01, 128.17, 129.43, 132.62, 133.87, 136.49, 144.19, 159.01 (phenolic), 196.99 (CO); mass: 253.3 [MH+], 275.2 [M + Na]+, 291.1 [M + K]+.
General procedure for the synthesis of partially reduced bisnaphthalenes (10a–f), bischromane (10g) and bisthiochromane (10h)
In a round-bottomed flask, compound 8 (3.95 mmol) was dissolved in dry dichloromethane. The contents of the flask were kept at about 0 °C on a salt bath for 20–30 minutes. Borontribromide (BBr3, 1.9 mL, 11.80 mmol) was then added to it, and the reaction mixture was kept at 0 °C for the next 30 minutes. The temperature of the reaction was increased to room temperature, and the progress of the reaction was monitored by TLC (ethyl acetate–hexane). After completion of the reaction, the reaction mixture was poured on ice and extracted with ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulphate (Na2SO4) and concentrated. The crude material was purified by column chromatography using silica gel (100–200 mesh size) and ethyl acetate–hexane as the eluent, which yielded pure compound 10.O); 1H NMR (CDCl3, 300 MHz, δ ppm): 2.37 (t, J = 14.4 Hz, 1H, CH of CH2), 2.68 (dd, J = 5.4 Hz & 14.7 Hz, 1H, CH of CH2), 3.24 (dd, J = 16.2 Hz & 13.2 Hz, 1H, CH of CH2), 3.43 (t, J = 12.3 Hz, 1H, CH of CH2), 3.64 (m, 2H, CH2), 3.78 (m, 7H, CH and 2xOCH3), 5.604 (s, 1H, CH), 6.80 (m, 6H, ArH), 6.98 (m, 3H, ArH), 7.05 (m, 3H, ArH), 7.31 (d, J = 7.2 Hz, 1H, ArH), 7.67 (m, 1H, ArH), 7.79 (m, 1H, CH), 8.21 (d, J = 7.5 Hz, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 37.31 (CH2), 38.21 (CH2), 40.05 (CH), 44.34 (CH), 55.17 (OCH3), 55.26 (OCH3), 62.51 (CH), 113.63 (2xAr–H), 113.74 (2xAr–H), 124.49 (Ar–H), 126.12 (Ar–H), 126.78 (Ar–H), 127.09 (Ar–H), 127.63 (Ar–H), 128.00 (Ar–H), 128.37 (2xAr–H), 128.46 (2xAr–H), 128.77 (Ar–H), 132.09, 132.23, 133.89 (2C), 134.83 (CH), 136.24 (Ar–H), 137.38, 142.98, 158.01 (phenolic), 158.42 (phenolic), 197.47 (CO); MS: 487 [MH+], 509 [M + Na]+, 525 [M + K]+; HRMS (ESI): calcd 487.2273 [MH+]; found: 487.2267; anal. calcd (C34H30O3): C, 83.92; H, 6.21; found: C, 83.80; H, 6.18%.O); 1H NMR (CDCl3, 300 MHz, δ ppm): 2.23 (m, 1H, CH of CH2), 2.62 (dd, J = 6.3 Hz and 15.0 Hz, 1H, CH of CH2), 3.15 (dd, J = 3.6 Hz and 15.9 Hz, 1H, CH of CH2), 3.39 (t, J = 12.0 Hz, 1H, CH of CH2), 3.63 (s, 3H, OCH3), 3.71 (m, 2H, 2xCH), 3.79 (dd, J = 3.6 Hz and 11.7 Hz, 1H, CH), 3.83 (s, 3H, OCH3), 5.64 (s, 1H, CH), 6.63 (s, 1H, ArH), 6.63 (d, J = 2.1 Hz, 1H, ArH), 6.83 (d, J = 6.6 Hz, 2H, ArH), 6.90 (d, J = 11.7 Hz, 1H, ArH), 6.97 (d, J = 20.7 Hz, 2H, ArH), 7.05–7.28 (m, 8H, Ar–H), 7.68 (s, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 36.69 (CH2), 37.65 (CH2), 41.82 (CH), 45.88 (CH), 55.52 (OCH3), 56.00 (OCH3), 62.50 (CH), 110.39 (Ar–H), 111.62 (Ar–H), 111.96 (Ar–H), 122.62 (Ar–H), 126.62 (Ar–H), 127.17 (Ar–H), 127.93 (2xAr–H), 128.01 (2xAr–H), 128.61 (Ar–H), 128.67 (2xAr–H), 128.87 (2xAr–H), 130.42 (Ar–H), 133.44, 133.81, 134.58, 135.00, 135.90, 136.94 (Ar–H), 143.16, 145.73, 158.31 (phenolic), 159.20 (phenolic), 197.70 (CO); MS: 487.4 [MH+]; HRMS (ESI): calcd 487.2273 [MH+]; found: 487.2256; anal. calcd (C34H30O3): C, 83.92; H, 6.21; found: C, 83.82; H, 6.17%.O); 1H NMR (CDCl3, 300 MHz, δ ppm): 2.32 (m, 3H, CH and CH2), 2.41 (m, 1H, CH of CH2), 2.75 (t, J = 8.1 Hz, 2H, CH2), 3.01 (m, 2H, CH2), 3.86 (t, J = 3.9 Hz 1H, CH), 5.80 (t, J = 4.5 Hz, 1H, CH), 7.08–7.15 (m, 4H, ArH), 7.26 (m, 1H, ArH), 7.34 (m, 1H, ArH), 7.50 (m, 1H, ArH), 8.10 (m, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 23.59 (CH2), 28.54 (CH2), 28.68 (CH2), 29.18 (CH2), 51.21 (CH), 123.54 (CH), 126.70 (Ar–H), 127.15 (Ar–H), 127.20 (Ar–H), 127.55 (Ar–H), 128.04 (Ar–H), 128.23 (Ar–H), 129.19 (Ar–H), 133.46, 133.82 (CH), 134.25, 135.72, 137.38, 144.48, 191.41 (CO); MS: 297 [M + Na]+; anal. calcd (C20H18O): C, 87.56; H, 6.61; found: C, 87.48; H, 6.58%.O); 1H NMR (CDCl3, 300 MHz, δ ppm): 2.11–2.38 (m, 4H, CH2 and 2xCH), 2.58–2.74 (m, 2H, CH2), 2.84–3.04 (m, 2H, 2xCH), 3.78 (s 3H, OCH3), 3.85 (m, 1H, CH), 3.92 (s, 3H, OCH3), 5.64 (t, J = 7.2 Hz, 1H, CH), 6.66–6.71 (m, 3H, ArH), 6.85 (dd, J = 2.4 Hz and 8.7 Hz, 1H, ArH), 7.01 (d, J = 8.1 Hz, 1H, ArH), 8.07 (d, J = 8.7 Hz, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 23.54 (CH2), 28.68 (CH2), 29.11 (CH2), 29.21 (CH2), 50.97 (CH), 55.62 (OCH3), 55.83 (OCH3), 111.29 (CH), 112.94 (Ar–H), 113.63 (Ar–H), 114.37 (Ar–H), 124.77 (Ar–H), 124.99 (Ar–H), 127.21, 127.38, 130.48 (Ar–H), 135.37, 139.28, 147.00, 158.73 (phenolic), 164.01 (phenolic), 198.30 (CO); MS: 335 [MH+]; anal. calcd (C22H22O3): C, 79.02; H, 6.63; found: C, 78.91; H, 6.57%.O); 1H NMR (CDCl3, 300 MHz, δ ppm): 2.23–2.27 (m, 3H, CH2), 2.32–2.43 (m, 1H, CH2), 2.60–2.71 (m, 2H, CH2), 2.83–3.022 (m, 2H, CH2), 3.73 (m, 7H, CH2 and 2xOCH3), 5.79 (t, J = 4.5 Hz, 1H, CH), 6.69 (t, 2H, J = 2.1 Hz, ArH), 7.07 (dd, J = 3.0 Hz and 8.4 Hz, 2H, ArH), 7.16 (d, J = 8.1 Hz, 1H, ArH), 7.58 (d, J = 2.4 Hz, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 23.95 (CH2), 27.54 (CH2), 27.73 (CH2), 29.37 (CH2), 50.84 (CH), 55.71 (OCH3), 55.91 (OCH3), 109.90 (Ar–H), 110.58 (Ar–H), 111.42 (CH), 122.21 (Ar–H), 128.11 (Ar–H), 128.72 (Ar–H), 129.57, 130.40 (Ar–H), 134.23, 135.36, 135.64, 137.09, 158.64 (phenolic), 158.84 (phenolic), 199.38 (CO); MS: 335.4 [MH+]; anal. calcd (C22H22O3): C, 79.02; H, 6.63; found: C, 78.93; H, 6.58%.O); 1H NMR (CDCl3, 300 MHz, δ ppm): 1.55 (s, 1H, CH), 2.18–2.38 (m, 4H, CH2), 2.66 (t, J = 7.8 Hz, 2H, CH2), 2.78–3.011 (m, 2H, CH2), 3.75 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 3.92 and 3.94 (s, 6H, 2xOCH3), 5.64 (t, J = 4.2 Hz, 1H, CH), 6.68, 6.703 & 6.736 (s, 3H, ArH), 7.59 (s, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 23.72 (CH2), 27.73 (CH2), 28.37 (CH2), 29.34 (CH2), 50.42 (CH), 56.37 (OCH3), 56.43 (OCH3), 56.46 (OCH3), 56.57 (OCH3), 108.52 (Ar–H), 109.25 (Ar–H), 110.62 (Ar–H), 112.08 (Ar–H), 125.22 (CH), 126.73, 127.05, 130.39, 135.38, 139.38, 147.54 (phenolic), 148.16 (phenolic), 148.51 (phenolic), 154.05 (phenolic), 198.42 (CO); MS: 395 [MH+]; anal. calcd (C24H26O5): C, 73.08; H, 6.64; found: C, 72.98; H, 6.59%.O); 1H NMR (acetone-d6, 300 MHz, δ ppm): 1.06 (s, 3H, CH3), 1.21 (s, 3H, CH3), 1.30 (s, 3H, CH3), 1.35 (s, 3H, CH3), 3.64 (s, 3H, OCH3), 3.68 (s, 1H, CH), 3.76 (s, 3H, OCH3), 5.27 (s, 1H, CH), 6.22 (d, 1H, J = 1.5 Hz, ArH), 6.37 (s, 2H, ArH), 6.49 (d, 1H, J = 1.2 Hz, ArH), 7.31 (d, 1H, J = 8.4 Hz, ArH), 7.61 (d, 1H, J = 8.7 Hz, ArH); 13C NMR (acetone-d6, 75 MHz, δ ppm): 24.37 (CH3), 26.36 (CH3), 26.84 (CH3), 27.23 (CH3), 55.10 (OCH3), 55.65 (OCH3), 75.74, 81.86, 101.64 (Ar–H), 102.72 (Ar–H), 106.75 (Ar–H), 109.80 (Ar–H), 114.20, 116.16, 125.16 (Ar–H), 127.92, 128.89 (CH), 155.03, 161.29 (phenolic), 161.63 (phenolic), 166.74, 190.04 (CO); MS: 395 [MH+]; anal. calcd (C24H26O5): C, 73.08; H, 6.64; found: C, 72.98; H, 6.59%.O); 1H NMR (CDCl3, 300 MHz, δ ppm): 3.150–3.29 (m, 2H, CH2), 3.36–3.54 (m, 2H, CH2), 4.09 (dd, J = 3.6 and 11.4 Hz, 1H, CH), 5.88 (t, J = 5.4 Hz, 1H, CH), 7.096–7.30 (m, 3H, ArH), 7.33–7.36 (m, 1H, ArH), 7.38–7.43 (m, 3H, ArH), 8.18 (dd, J = 1.5 and 8.1 Hz, 1H, Ar–H); 13C NMR (CDCl3, 75 MHz, δ ppm): 25.26 (CH2), 32.02 (CH2), 52.48 (CH), 122.55 (CH), 125.54 (2xAr–H), 126.14 (Ar–H), 128.02 (Ar–H), 128.10 (Ar–H), 128.58 (Ar–H), 130.28 (Ar–H), 131.71, 133.03, 133.77 (Ar–H), 134.62, 136.59, 142.39, 194.87 (CO); MS: 311.2 [MH+]; HRMS (ESI): calcd 311.0564 [MH+]; found: 311.0546; anal. calcd (C18H14OS2): C, 69.64; H, 4.55; found: C, 69.54; H, 4.51%.General procedure for the synthesis of partially reduced bisindene (12)
Compound 11 (0.59 mmol) was treated with borontribromide (BBr3, 0.30 mL, 1.76 mmol) under similar reaction conditions to those discussed above for 10. The crude material obtained from the reaction was purified by column chromatography using silica gel (100–200 mesh size) and ethyl acetate–hexane (4:96) as the eluent, which yielded pure compound 12.
O); 1H NMR (CDCl3, 300 MHz, δ ppm): 0.88 (t, 1H, J = 6.6 Hz, CH of CH2), 1.25 (t, 1H, J = 6.9 Hz, CH of CH2), 3.74 (s, 3H, OMe), 3.78 (s, 3H, OMe), 4.46 (t, 1H, J = 4.2 Hz, CH), 5.36 (s, 1H, CH), 6.80–6.84 (m, 3H, ArH), 6.98–7.15 (m, 4H, ArH), 7.23–7.39 (m, 7H, ArH), 7.42–7.57 (m, 1H, ArH), 7.81 (d, 1H, J = 7.8, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 43.64 (CH2), 48.89 (CH), 49.03 (CH), 55.58 (OCH3), 55.67 (OCH3), 114.41 (2xAr–H), 114.70 (Ar–H), 114.92 (Ar–H), 124.05 (Ar–H), 125.10, 125.91 (Ar–H), 126.30 (Ar–H), 127.38 (Ar–H), 127.62 (Ar–H), 127.88 (Ar–H), 128.66 (2xAr–H), 129.07 (2xAr–H), 129.96, 131.12 (Ar–H), 134.57 (Ar–H), 135.33, 136.64, 137.69, 138.00, 139.65, 156.22, 158.82, 196.11 (CO); MS: 457 [M − H]+; anal. calcd (C32H26O3): C, 83.82; H, 5.72; found: C, 83.70; H, 5.66%.O); 1H NMR (CDCl3, 300 MHz, δ ppm): 3.09 (d, J = 5.1 Hz, 2H, CH2), 3.58 (d, J = 5.1 Hz, 2H, CH2), 3.96 & 4.01 (s, 14H, 4xOCH3 and CH2), 6.92 (s, 1H, ArH), 7.01 (s, 1H, ArH), 7.28 (s, 1H, ArH), 7.30 (s, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 31.33 (CH2), 32.47 (CH2), 32.72 (CH2), 56.38 (OCH3), 56.49 (OCH3), 56.58 (OCH3), 56.66 (OCH3), 104.98 (Ar–H), 107.51 (Ar–H), 108.05 (Ar–H), 108.42 (Ar–H), 124.75, 133.43, 133.58, 143.23, 146.32, 148.98, 149.77 (phenolic), 152.24 (phenolic), 154.48 (phenolic), 154.74 (phenolic), 194.36 (CO); MS: 367 [M + H]+; HRMS (ESI): calcd 367.1545 [MH+]; found: 367.1531; anal. calcd (C22H22O5): C, 72.12; H, 6.05; found: C, 72.01; H, 6.01%.O); 1H NMR (CDCl3, 300 MHz, δ ppm): 3.09 (t, J = 5.7 Hz, 2H, CH2), 3.53 (t, J = 5.7 Hz, 2H, CH2), 3.86 and 3.90 (s, 8H, 2xOCH3 and CH2), 6.87–6.96 (m, 4H, ArH), 7.66 (d, J = 8.4 Hz, 1H, ArH), 7.76 (d, J = 8.4 Hz, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 31.47 (CH2), 32.15 (CH2), 33.40 (CH2), 55.83 (OCH3), 55.94 (OCH3), 109.89 (Ar–H), 110.20 (Ar–H), 114.48 (Ar–H), 115.00 (Ar–H), 124.90, 125.50 (Ar–H), 127.33 (Ar–H), 133.75, 134.20, 151.51, 153.83, 154.43, 162.13 (phenolic), 164.63 (phenolic), 194.15 (CO); MS: 307.3 [MH+]; anal. calcd (C20H18O3): C, 78.41; H, 5.92; found: C, 78.31; H, 5.89%.O); 1H NMR (CDCl3, 300 MHz, δ ppm): 3.07 (t, J = 5.7 Hz, 2H, CH2), 3.57 (t, J = 5.7 Hz, 2H, CH2), 3.86–3.97 (m, 8H, 2xOCH3 and CH2), 6.97 (d, J = 8.1 Hz, 1H, ArH), 7.17 (dd, J = 3.2 and 5.7 Hz, 1H, ArH), 7.25 (s, 1H, ArH), 7.31 (s, 1H, ArH), 7.33 (s, 1H, ArH), 7.41 (d, J = 8.1 Hz, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 30.47 (CH2), 32.53 (CH2), 32.70 (CH2), 55.94 (OCH3), 56.04 (OCH3), 105.67 (Ar–H), 111.18 (Ar–H), 117.26 (Ar–H), 123.11 (Ar–H), 126.47 (Ar–H), 127.06 (Ar–H), 127.56, 141.10, 141.53, 142.43, 144.49, 155.43, 159.23 (phenolic), 159.81 (phenolic), 195.43 (CO); MS: 307.3 [MH+]; anal. calcd (C20H18O3): C, 78.41; H, 5.92; found: C, 78.29; H, 5.88%.O); 1H NMR (pyridine-d5, 300 MHz, δ ppm): 2.90 (t, J = 5.7 Hz, 2H, CH2), 3.53 (t, J = 5.7 Hz, 2H, CH2), 3.83 (s, 2H, CH2), 7.54 (s, 1H, ArH), 7.58 (m, 3H, ArH), 7.74 (s, 1H, ArH), 7.78 (d, J = 8.1 Hz, 1H, ArH); MS: 405.1 [MH+]; anal. calcd (C18H12Br2O): C, 53.50; H, 2.99; found: C, 53.39; H, 2.96%.O); 1H NMR (CDCl3, 300 MHz, δ ppm): 2.27 (s, 3H, CH3), 2.32 (s, 3H, CH3), 2.94 (t, J = 5.7 Hz, 2H, CH2), 3.40 (t, J = 5.7 Hz, 2H, CH2), 3.78 (s, 2H, CH2), 7.00–7.09 (m, 3H, ArH), 7.13 (s, 1H, ArH), 7.58 (d, J = 8.1 Hz, 1H, ArH), 7.51 (d, J = 7.8 Hz, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 22.03 (CH3), 22.52 (CH3), 31.16 (CH2), 32.05 (CH2), 33.27 (CH2), 123.81 (Ar–H), 126.08 (Ar–H), 126.20, 126.66 (Ar–H), 126.73 (Ar–H), 128.35 (Ar–H), 128.84 (Ar–H), 137.93, 138.79, 141.33, 144.81, 149.39, 152.38, 154.89, 195.27 (CO); MS: 275.4 [MH+]; anal. calcd (C20H18O): C, 87.56; H, 6.61; found: C, 87.43; H, 6.58%.General procedure for the synthesis of 2,3-dihydro-5-methoxy-3-(4-methoxyphenyl)-2-phenylinden-1-one (15)
In a round-bottomed flask, 2,3-dihydro-5-methoxy-3-(4-hydroxyphenyl)-2-phenylinden-1-one (14) (0.25 g, 0.76 mmol) was dissolved in dry acetone. Anhydrous potassium carbonate (0.84 g, 6.06 mmol) was added to this solution. After a few minutes of stirring at room temperature, dry dimethyl sulphate (0.22 mL, 2.27 mmol) was added to the reaction mixture. The reaction mixture was further stirred at room temperature for 1.5 h. The progress of the reaction was monitored by TLC using 10% ethyl acetate–benzene. The formation of compound was completed in 1.5 h. After completion of the reaction, the solvent was distilled, and the reaction mixture was worked up with ethyl acetate–water. The organic layer was separated, dried over anhydrous sodium sulphate (Na2SO4) and concentrated. The crude material was purified by crystallization using acetone–hexane (3:97), which yielded pure compound 15 as a white solid in 85% yield; Rf: 0.64 (10% ethyl acetate–benzene); mp: 107–108 °C; IR (KBr, νmax/cm−1): 1700 (CO); 1H NMR (CDCl3, 300 MHz, δ ppm): 3.66 (d, J = 4.5 Hz, 1H, CH), 3.76 (bs, 6H, 2xOCH3), 4.36 (d, J = 4.2 Hz, 1H, CH), 6.61 (s, 1H, CH), 6.79 (d, J = 6.6 Hz, 2H, ArH), 6.92–6.95 (m, 3H, ArH), 7.01 (d, J = 6.6 Hz, 2H, ArH), 7.18–7.24 (m, 3H, ArH), 7.7 (d, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 54.71 (OCH3), 55.68 (OCH3), 56.13 (CH), 65.35 (CH), 109.95 (Ar–H), 114.71 (2xAr–H), 116.80 (Ar–H), 126.13 (Ar–H), 127.45 (Ar–H), 128.69 (2xAr–H), 129.20 (2xAr–H), 129.32 (2xAr–H), 129.96, 134.98, 139.47, 159.13 (phenolic), 159.87 (phenolic), 166.30, 203.88 (CO); MS: 345 [MH+]; anal. calcd (C23H20O3): C, 80.21; H, 5.85; found: C, 80.11; H, 5.83%.
General procedure for the synthesis of 2,3-dihydro-5-hydroxy-3-(4-methoxyphenyl)-2-phenylinden-1-one (16)
Compound 15 (0.05 g, 0.15 mmol) was treated with borontribromide (BBr3, 0.10 mL, 0.43 mmol) under similar reaction conditions to those discussed above for 10. The purification of crude oily material obtained from the reaction was made by column chromatography using silica gel (100–200 mesh size) and benzene as the eluent and yielded pure compound 16 as a white solid in 82% yield; Rf: 0.29 (10% ethyl acetate–benzene); mp: 213–214 °C; IR (KBr, νmax/cm−1): 1679 (CO); 1H NMR (acetone-D6, 300 MHz, δ ppm): 3.80 (m, 4H, OCH3 and CH), 4.511 (bs, 1H, CH), 6.80 (m, 3H, ArH), 7.05 (m, 5H, ArH), 7.30 (m, 3H, ArH), 7.68 (m, 1H, ArH), 8.40 (bs, 1H, OH); 13C NMR (acetone-D6, 100 MHz, δ ppm): 54.08 (CH), 55.73 (OCH3), 65.02 (CH), 110.12, 116.01 (2xC), 116.40, 125.40, 127.12, 128.78 (3xC), 129.33 (2xC), 129.53 (2xC), 133.77, 139.91, 156.95 (phenolic), 160.09 (phenolic), 166.22, 202.39 (CO); MS: 330 [M]+, 331 [MH+]; anal. calcd (C22H18O3): C, 79.98; H, 5.49; found: C, 79.89; H, 5.44%.
7,7′-Dimethyl-2′,3′-dihydro-1′H,3H-1,2′-biinden-1′-one (17)
The synthesis was carried out following the procedure as reported for 12. The crude material obtained from the reaction was purified by column chromatography using silica gel (100–200 mesh size) and ethyl acetate–hexane (4:96) as the eluent, which yielded pure compound 17. Off-white solid; yield: 86%; Rf: 0.60 (20% ethyl acetate–hexane); mp: 123–113 °C; IR (KBr, νmax/cm−1): 1702 (CO); 1H NMR (CDCl3, 300 MHz, δ ppm): 2.53 (s, 3H, CH3), 2.57 (s, 3H, CH3), 3.03–3.18 (m, 3H, CH2 & CH of CH2), 3.51 (p, J = 8.4 Hz, 1H, CH of CH2), 4.16 (t, J = 4.8 Hz, 1H, CH), 6.02 (s, 1H, CH), 6.94–7.05 (m, 3H, ArH), 7.13–7.18 (m, 2H, ArH), 7.33 (d, J = 7.5 Hz, 1H, ArH); 13C NMR (CDCl3, 75 MHz, δ ppm): 18.81 (CH3), 20.65 (CH3), 35.81 (CH2), 38.01 (CH2), 49.02 (CH), 122.21 (Ar–H), 124.31 (Ar–H), 125.26 (Ar–H), 129.75 (Ar–H), 129.89 (Ar–H), 130.19, 130.91, 134.42, 134.69 (CH), 139.83, 142.63, 144.72, 145.65, 154.09, 207.38 (CO); MS: 275.4 [MH+]; anal. calcd (C20H18O): C, 87.56; H, 6.61; found: C, 87.40; H, 6.57%.
Procedure for crossed reactions of tetralones (8d and 8f)
In a round-bottomed flask, compounds 8d (170 mg, 0.96 mmol) and 8f (200 mg, 0.96 mmol) were dissolved in dry dichloromethane. The mixture was kept at about 0 °C on a salt bath for 20–30 minutes. Borontribromide (BBr3, 0.97 mL, 5.8 mmol) was then added to it, and the reaction mixture was kept at 0 °C for the next 30 minutes. The temperature of the reaction was brought to room temperature, and the progress of the reaction was monitored by TLC (acetone–hexane). After completion of the reaction (5 h), the reaction mixture was poured on ice and extracted with ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulphate (Na2SO4) and concentrated. The crude material was purified by flash chromatography using silica gel (200–400 mesh size) and acetone–hexane (1:49) as the eluent, which yielded pure compounds 10d (25%) and 10f (26%) along with an inseparable mixture of 18 (18a + 18b, 27%).
Mixture of 6,6′,7′-trimethoxy-3,3′,4,4′-tetrahydro-1,2′-binaphthyl-1′-(2′H)-one (18a)a and 6,6′,7-trimethoxy-3,3′,4,4′-tetrahydro-1,2′-binaphthyl-1′-(2′H)-one (18b)b
Off white solid; yield: 27%; Rf: 0.28 (20% acetone–hexane); mp: 148–149 °C; IR (KBr, νmax/cm−1): 1665 (CO); 1H NMR (CDCl3, 300 MHz, δ ppm): 35% (18a)a + 65% (18b)b, 2.15–2.37 (m, 8H, CH2a,b), 2.63–2.83 (m, 4H, CH2a,b), 2.83–2.98 (m, 4H, CH2a,b), 3.72–3.93 (m, 20H, OMea,b and CHa,b), 5.60–5.68 (m, 2H, CHa,b), 6.65–6.71 (m, 6H, ArHa,b), 6.85 (d, J = 11.2 Hz, 1H, ArHb), 7.03 (d, J = 10.8 Hz, 1H, ArHa), 7.59 (s, 1H, ArHa), 8.07 (d, J = 11.6 Hz, 1H, ArHb); 13C NMR (CDCl3, 75 MHz, δ ppm): 23.53, 23.73, 27.96, 28.38, 28.55, 29.16, 29.35, 30.07, 31.27, 51.00, 51.07, 55.61, 55.83, 56.37, 56.63, 108.61, 109.32, 110.61, 111.29, 112.08, 112.95, 113.66, 114.37, 124.71, 125.42, 126.98, 127.18, 130.41, 135.47, 146.98, 147.52, 148.13, 164.06, 198.27; MS: 365 [MH+].
Procedure for crossed reactions of indanones (11b and 11c)
In a round-bottomed flask, compounds 11b (200 mg, 1.04 mmol) and 11c (168 mg, 1.04 mmol) were dissolved in dry dichloromethane. The mixture was kept at about 0 °C on a salt bath for 20–30 minutes. Borontribromide (BBr3, 1.0 mL, 6.2 mmol) was then added to it, and the reaction mixture was kept at 0 °C for the next 30 minutes. The temperature of the reaction was brought to room temperature, and the progress of the reaction was monitored by TLC (ethyl acetate–hexane). After completion of the reaction (5 h), the reaction mixture was poured on ice and extracted with ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulphate (Na2SO4) and concentrated. The crude material was purified by flash chromatography using silica gel (200–400 mesh size) and acetone–hexane (1:49) as the eluent, which yielded pure compounds 12b (29%) and 12c (30%) along with an inseparable mixture of 19 (19a + 19b, 31%).
Mixture of 5,6-dimethoxy-2-(5-methoxy-2,3-dihydro-1H-inden-1-ylidene)-2,3-dihydro-1H-inden-1-one (19a)a and (2Z)-2-(5,6-dimethoxy-2,3-dihydro-1H-inden-1-ylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (19b)b
Yellow solid; yield: 31%; Rf: 0.20 (20% acetone–hexane); mp: 172–173 °C; IR (KBr, νmax/cm−1): 1670 (CO); 1H NMR (CDCl3, 300 MHz, δ ppm): 30% (19a)a + 70% (19b)b, 2.99–3.06 (m, 4H, CH2a,b), 3.48–3.50 (m, 4H, CH2a,b), 3.79–3.96 (m, 22H, OMea,b & CHa,b), 6.82–6.93 (m, 6H, ArHa,b), 7.18–7.25 (m, 2H, ArHa,b), 7.61 (d, J = 11.6 Hz, 1H, ArHa), 7.72 (d, J = 11.2 Hz, 1H, ArHb); 13C NMR (CDCl3, 75 MHz, δ ppm): 30.07, 31.26, 31.45, 32.11, 32.46, 33.04, 55.82, 55.93, 56.35, 56.44, 56.54, 56.68, 104.91, 107.49, 108.45, 109.81, 110.18, 114.47, 115.13, 124.43, 125.46, 127.26, 133.54, 133.77, 134.20, 143.50, 146.34, 148.95, 151.23, 152.24, 153.44, 154.37, 154.75, 162.11, 164.62, 194.02, 207.21; MS: 337 [MH+].
Procedure for crossed reactions of tetralone (8d) and indanone (11c)
In a round-bottomed flask, compounds 8d (200 mg, 1.13 mmol) and 11c (184 mg, 1.13 mmol) were dissolved in dry dichloromethane. The mixture was kept at about 0 °C on a salt bath for 20–30 minutes. Borontribromide (BBr3, 1.1 mL, 6.8 mmol) was then added to it, and the reaction mixture was kept at 0 °C for the next 30 minutes. The temperature of the reaction was brought to room temperature, and the progress of the reaction was monitored by TLC (ethyl acetate–hexane). After completion of the reaction (6 h), the reaction mixture was poured on ice and extracted with ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulphate (Na2SO4) and concentrated. The crude material was purified by flash chromatography using silica gel (200–400 mesh size) and acetone–hexane (1:49) as the eluent, which yielded pure compounds 10d (37%) and 12c (33%).
Procedure for crossed reactions of tetralone (8d) and indanone (11g)
In a round-bottomed flask, compounds 8d (240 mg, 1.36 mmol) and 11g (200 mg, 1.36 mmol) were dissolved in dry dichloromethane. The mixture was kept at about 0 °C on a salt bath for 20–30 minutes. Borontribromide (BBr3, 1.3 mL, 8.2 mmol) was then added to it, and the reaction mixture was kept at 0 °C for next 30 minutes. The temperature of the reaction was brought to room temperature, and the progress of the reaction was monitored by TLC (ethyl acetate–hexane). After completion of the reaction (6 h), the reaction mixture was poured on ice and extracted with ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulphate (Na2SO4) and concentrated. The crude material was purified by flash chromatography using silica gel (200–400 mesh size) and acetone–hexane (1:49) as the eluent, which yielded pure compounds 10d (18%), 17 (27%), 20 (23%) and 21 (20%).
2-(6-Methoxy-3,4-dihydronaphthalen-1-yl)-7-methyl-2,3-dihydro-1H-inden-1-one (20)
Pale yellow oily product; yield: 23%; Rf: 0.78 (20% acetone–hexane); IR (KBr, νmax/cm−1): 1675 (CO); 1H NMR (CDCl3, 400 MHz, δ ppm): 2.28–2.33 (m, 2H, CH2), 2.71 (s, 3H, CH3), 2.73–2.79 (m, 2H, CH2), 3.13 (dd, J = 17.32 and 4.50 Hz, 1H, CH2), 3.50 (q, J = 8.4 Hz, 1H, CH2), 3.78–3.79 (m, 1H, CH), 3.80 (s, 3H, OMe), 5.83 (t, J = 4.8 Hz, 1H, CH), 6.68 (dd, J = 8.48 and 2.40 Hz, 1H, ArH), 6.74 (d, J = 2.6 Hz, 1H, ArH), 6.89 (d, J = 7.60 and 3.60 Hz, 1H, ArH), 7.18 (d, J = 7.4 Hz, 1H, ArH), 7.29 (dd, J = 7.6 and 3.6 Hz, 1H, ArH), 7.50 (t, J = 7.48 Hz, 1H, ArH); 13C NMR (CDCl3, 100 MHz, δ ppm): 18.39 (CH3), 23.18 (CH2), 28.70 (CH2), 33.90 (CH2), 51.73 (CH), 55.21 (OCH3), 110.86, 114.15, 123.92, 123.95, 125.23, 126.93, 129.39, 134.16, 134.30, 135.25, 138.83, 139.37, 154.05, 158.50, 207.40 (CO); MS: 305 [MH+]; anal. calcd (C21H20O2): C, 82.86; H, 6.62; found: C, 82.73; H, 6.58%.
6-Methoxy-2-(4-methyl-1H-inden-3-yl)-3,4-dihydronaphthalen-1(2H)-one (21)
Pale yellow solid; yield: 20%; Rf: 0.68 (20% acetone–hexane); mp: 112–113 °C; IR (KBr, νmax/cm−1): 1675 (CO); 1H NMR (CDCl3, 400 MHz, δ ppm): 2.40–2.47 (m, 2H, CH2), 2.56 (s, 3H, CH3), 3.00–3.06 (m, 2H, CH2), 3.35 (bs, 2H, CH2), 3.90 (s, 3H, OMe), 4.24 (t, J = 5.6 Hz, 1H, CH), 6.17 (bs, 1H, CH), 6.76 (s, 1H, ArH), 6.91 (t, J = 2.4 Hz, 1H, ArH), 7.07 (d, 1H, ArH), 7.12 (t, J = 7.2 Hz, 1H, ArH), 7.32 (d, 1H, ArH), 8.12 (d, J = 8.8 Hz, 1H, ArH); 13C NMR (CDCl3, 100 MHz, δ ppm): 20.09 (CH3), 28.12 (CH2), 29.44 (CH2), 37.78 (CH2), 48.45 (CH), 55.47 (OCH3), 112.55, 113.31, 121.80, 124.71, 126.62, 129.43, 130.04, 130.20, 130.50, 142.55, 143.81, 144.98, 146.60, 163.65, 197.35 (CO); MS: 305 [MH+]; anal. calcd (C21H20O2): C, 82.86; H, 6.62; found: C, 82.75; H, 6.59%.
Crystallography
with one molecule in the asymmetric unit. a = 9.957 (1) Å, b = 10.296 (2) Å, c = 13.293 (2) Å, α = 74.756° (4), β = 76.427°, γ = 79.167°, Z = 2, V = 1266.4 (3) Å3, Dcalc = 1.276 g cm−3. X-ray diffraction data were collected on a Bruker AXS SMART APEX CCD diffractometer using MoKα radiation (λ = 0.71073 Å). Data were acquired using the ω scan mode at room temperature (293 K). A total of 11707 reflections were measured with 6347 unique reflections (Rint = 0.055). The structure was solved by direct methods using SHELXS and was refined against F2 with full-matrix least squares method by using SHELXL. All the nonhydrogen atoms were refined anisotropically. Moreover, all the H-atoms, except those of the methoxy group, were located from the differences in Fourier maps and were refined isotropically. Methoxy hydrogens were fixed geometrically in idealized positions and were refined using a riding atom model. The final R-factor was 0.0629 and wR = 0.1163 for 3239 observed reflections with [I > 2sigI]. The goodness-of-fit was 0.939. with one molecule in the asymmetric unit. The unit cell dimensions were a = 7.952 (3) Å, b = 9.721 (4) Å, c = 9.932 (4) Å, α = 109.057° (7), β = 102.134° (7), γ = 92.430° (7), Z = 2, V = 704.3 (5) Å3, Dcalc = 1.294 g cm−3. Data were acquired using the ω scan mode at room temperature (293 K). The structure was solved by direct methods using SHELXS and was refined against F2 with full-matrix least squares method by using SHELXL. All nonhydrogen atoms were refined anisotropically. Moreover, all the H-atoms, except those of the methyl groups, were located from the differences in Fourier maps and were refined isotropically. Methyl hydrogens were fixed geometrically in idealized positions and were refined using a riding atom model. A total of 3617 reflections were measured with 2382 unique reflections (Rint = 0.019). The final R was 0.0574 and wR = 0.1522 for 1866 observed reflections with [I > 2sigI]. The goodness-of-fit was 1.075.718 reflections were measured with 1984 unique reflections (Rint = 0.27). The final R was 0.0885 and wR = 0.1698 for 691 observed reflections with [I > 2sigI]. The goodness-of-fit was 0.975.Acknowledgements
A.G. and H.K.M. acknowledge DST, New Delhi, India for financial support, and I.A. acknowledges CSIR, New Delhi, India for financial support as SRF. The X-ray diffraction facility at the CESE, IIT Kanpur is acknowledged. Support received from the Director, CSIR-CIMAP, India is duly acknowledged.Notes and references
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS; (b) S. G. Modha, V. P. Mehta and E. V. V. der Eycken, Chem. Soc. Rev., 2013, 42, 5042–5055 RSC; (c) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC.
- V. P. Mehta and B. Punji, RSC Adv., 2013, 3, 11957–11986 RSC.
- E. G. Doyagüez, Synlett, 2005, 1636–1637 CrossRef.
- Y. Komatsu, V. D. Mihailetchi, L. J. Geerligs, B. van Dijk, J. B. Rem and M. Harris, Sol. Energy Mater. Sol. Cells, 2009, 93, 750–752 CrossRef CAS.
- S. Ray and I. Dwivedy, Adv. Drug Res., 1997, 29, 171–270 CAS.
- (a) L. M. Rasmussen, N. T. Zaveri, J. Stenvang, R. H. Peters and A. E. Lykkesfeldt, Breast Cancer Res. Treat., 2007, 106, 1910–1916 CrossRef; (b) M. Tanabe, R. H. Peters, W. Chao and L. Jong, US 20020032180 A1, 2002.
- The 1H NMR spectra of 4 showed two sharp singlets at 4.51 ppm for two protons and at 6.25 ppm for one proton, which indicated the presence of one allylic CH2 attached to an ester group and one olefinic CH attached to a phenyl group. Similarly, 1H NMR spectra of compound 5 showed two singlets at 3.69 ppm for one CH2 and at 6.97 ppm for CH, which indicated the presence of one CH2 attached to a phenyl ring (benzylic CH2) and one CH group attached to an ester group (conjugated CH).
- It is worthwhile to mention that on silica gel TLC, compounds 4 and 6 had Rf values of 0.34 and 0.27, respectively (in a 10% ethyl acetate–hexane solvent system) and could be identified easily, whereas compounds 5 and 6 had almost the same Rf values (0.27 and 0.26 in 10 % ethyl acetate–hexane) on TLC. Therefore, the reaction could not be monitored for product formation even after using different combinations of solvents. In such cases, the product formation could only be confirmed through 1H NMR spectroscopy. In 1H NMR spectra, compound 6 showed the presence of two multiplets at 2.59 and 2.93 ppm for two CH2 along with one multiplet at 3.36 ppm for one CH, which indicated reduction of the CC bond and formation of product 6. Disappearance of olefinic singlets from 1H NMR spectra of 4 and 5 further confirmed the formation of compound 6.
-
(a) Methanol and water (1:1) were used for HPLC analysis of 10a;
(b) The Rf value of 10a (0.45 in 20% ethyl acetate–hexane) on TLC was much higher than that of the authentic hydroxy derivative (compound 9, Rf value 0.25 in 20% ethyl acetate–hexane) and very close to that of the starting compound (8a, Rf value 0.57). In infrared (IR) spectrum, compound 10a showed bands at 3001, 2935, and 2834 cm−1, indicating the presence of a number of CH2 and CH groups; moreover, a band at 1678 cm−1 indicated the presence of one carbonyl (CO) functional group in the compound. Furthermore, product (10a) showed peaks at 487, 509 and 525 in its mass spectrum, which indicated that the molecular weight of 10a could be between 486 and 526. The 1H NMR of 10a had different signals for 30 protons, whereas signals in 13C NMR showed the presence of seven C-atoms in the aliphatic region (between 20–80 ppm), including two signals for two methoxy groups. In an aromatic region (between 80–200 ppm), signals were present for 27 C-atoms, including one for a carbonyl (CO) group. From the above information, it could be deduced that compound 10a had a total of 30 protons, 34 C-atoms and 3 oxygen atoms (1 from a carbonyl group and 2 from methoxy groups). Thus, the molecular formula of 10a could be C34H30O3 with a molecular weight of 486, which was within the observed range (as in mass spectra). The double bond equivalents calculated for 10a as per this molecular formula were 20, which later accounted for 14 double bonds and 6 rings present in the molecule (Scheme 2). The spectral analysis of 1H NMR further showed the presence of a triplet and double doublet for two CH groups at 2.36 and 2.68 ppm, respectively, indicating that these signals were for H5 and H6 (germinal protons), which were attached to the C4. This was clearly observed in HSQC spectra (same C-signal for both protons). Another set of a triplet and double doublet for two CH groups at 3.43 and 3.23 ppm was for H15 and H16, respectively (Scheme 2). These protons were also attached to the same C-atom (C-4′) as shown in the HSQC spectra. The interactions between H5 and H6 as shown in the COSY spectra further revealed that these protons were in close proximity. Similarly, H15 and H16 showed interactions in COSY spectra, and therefore, these were in close proximity. Furthermore, in COSY spectra interactions between H5 and H6 with one proton that appeared at around 3.6–3.8 ppm showed its proximity to them; therefore, it could be assigned as H7. In 1H NMR, a singlet that appeared at 5.60 ppm for one proton could be assigned as a olefinic (HCC) proton, which showed its interaction with one of the protons that appeared at around 3.6–3.8 ppm (one of the CH) in the COSY spectra. This showed its proximity with H15; therefore, it could be assigned as H14. The presence of this olefinic proton was further confirmed by 13C NMR, which showed a signal at 124.49 ppm. This information also was supported by HSQC spectra. The presence of two methoxy groups in the molecule could be explained by the presence of a multiplet at 3.78 ppm for seven protons (6 protons for two OCH3 groups and one CH) in the 1H NMR spectra and two signals at 55.17 and 55.26 ppm for methoxy groups and two signals at 158.01 and 158.42 ppm for phenolic C-atoms in 13C NMR spectra. One doublet at 8.21 ppm with a coupling constant (J) of 7.5 Hz in 1H NMR spectra showed the presence of one ortho-coupled peri-proton (H1). In the COSY spectra, this proton showed interactions with a proton appearing at 7.67 ppm, indicating its proximity to it. Therefore, it could be assigned later as H2. The DEPT spectra revealed the presence of a total of 17 CH groups and 10 quaternary C-atoms in the
aromatic region, while two CH2 groups, two methoxy groups and two CH groups were found in the aliphatic region. Thus, the structure of this compound was assigned as partially reduced bisnaphthalene (10a).
- J. Muzart, Synth. Commun., 1985, 15, 285–289 CrossRef CAS.
- J. W. Barlow, A. P. McHugh, O. Woods and J. J. Walsh, Eur. J. Med. Chem., 2011, 46, 1545–1554 CrossRef CAS.
- A. G. Holba, V. Premasager, B. C. Barot and E. J. Eisenbraun, Tetrahedron Lett., 1985, 26, 571–574 CrossRef CAS.
- S. Hagen and L. T. Scott, J. Org. Chem., 1996, 61, 7198–7199 CrossRef CAS.
- J. W. Burnham, R. G. Melton, E. J. Eisenbraun, G. W. Keen and M. C. Hamming, J. Org. Chem., 1973, 38, 2783–2785 CrossRef CAS.
- M. Orchin, L. Reggel and R. A. Friedel, J. Am. Chem. Soc., 1949, 71, 2743–2745 CrossRef CAS.
- H.-Y. Yin, M. Y. Liu and L. X. Shao, Org. Lett., 2013, 15, 6042–6045 CrossRef.
- (a) Y. Yang, D. Philips and S. Pan, J. Org. Chem., 2011, 76, 1902–1905 CrossRef CAS; (b) W. Denny, R. H. Hutchings, D. S. Johnson, J. S. Kaltenbronn, H. H. Lee, D. M. Leonard, J. B. J. Milbank, J. T. Repine, G. W. Rewcastle and A. D. White, WO 2001079180, 2001.
- (a) S. A. Snyder, S. P. Breazzano, A. G. Ross, Y. Lin and A. L. Zografos, J. Am. Chem. Soc., 2009, 131, 1753–1765 CrossRef CAS; (b) W. Skuballa, B. Raduechel, O. Loge, W. Elger and H. Vorbrueggen, J. Med. Chem., 1978, 21, 443–447 CrossRef CAS; (c) O. W. Woltersdorf Jr, S. J. DeSolms, E. M. Schultz and E. J. Cragoe Jr, J. Med. Chem., 1977, 20, 1400 CrossRef CAS; (d) H. E. Zimmerman and P. H. Lamers, J. Org. Chem., 1989, 54, 5788–5804 CrossRef CAS.
- ESI.†.
- (a) C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC; (b) F.-G. Klärner and B. Kahlert, Acc. Chem. Res., 2003, 36, 919 CrossRef; (c) M. Harmata, Acc. Chem. Res., 2004, 37, 862 CrossRef CAS.
- (a) V. Percec, M. Glodde, T. K. Bera, Y. Miura, I. Shiyanovskaya, K. D. Singer, V. S. K. Balagurusamy, P. A. Heiney, I. Schnell, A. Rapp, H.-W. Spiess, S. D. Hudson and H. Duan, Nature, 2002, 417, 384 CrossRef; (b) M. R. Bryce, Adv. Mater., 1999, 11, 11 CrossRef CAS; (c) G. W. Coates, A. R. Dunn, L. M. Henling, D. A. Doughtery and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1997, 36, 248 CrossRef CAS.
- A. A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 938650 (10a) and 980068 (17). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra01745e |
| This journal is © The Royal Society of Chemistry 2014 |