
A convenient synthesis of pyrimidinone and pyrimidine containing bisheteroarenes and analogs
Abstract
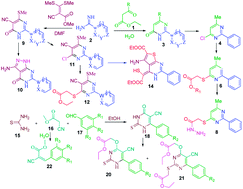
The synthesis of pyrimidinone containing bisheteroarenes (3) and related analogs (9 and 10) by the reaction of active methylenes or substituted methyl acrylate with nitrogen containing precursors viz. amidines, or thiourea in water as well as other organic solvents was studied. Synthesized compounds have further been explored for the synthesis of diversified pyrimidines 4, 6–8, 11, 12 and 14 through a sequential approach.
 Please wait while we load your content...
Please wait while we load your content...

