DOI:
10.1039/C4RA01678E
(Paper)
RSC Adv., 2014,
4, 25719-25728
Matrix effects caused by phospholipids in multi-residue analysis for beta-agonists with liquid chromatography-electrospray tandem mass spectrometry
Received
26th February 2014
, Accepted 21st May 2014
First published on 23rd May 2014
Abstract
It was first confirmed that 5 glycerophospholipids (GPs, (15.6 mg L−1)), including lysophosphatidylcholine (LPC C18:0), lysophosphatidylcholine, LPC (C16:0), phatidylcholine, PC (C16:0/C18:2), phatidylcholine, PC (C16:0/C18:1) and phatidylethanolamine, PE (C18:0/C20:4), could significantly suppress the ionization of nine beta-agonists (2 μg L−1) using post-column infusion during liquid chromatography-electrospray tandem mass spectrometry analyses. Under optimal experimental conditions, most beta-agonists such as fenoterol, clorprenaline, tulobuterol, clenbuterol and especially penbuterol were co-eluted with GPs, and a positive linear correlation (R2 > 0.85) between the amount of GPs and the extent of ionization suppression was observed for the five analytes. In addition, the linear correlation was statistically significant (p < 0.001). GPs including LPC C18:0 (m/z 496.2), PC C16:0/C18:2 (m/z 758.5), PC C16:0/C18:1 (m/z 760.5), PE C18:0/C20:4 (m/z 768.6) and plasmalogen phosphatidylcholine, PLPC C18 (Plasm)/C18:1 (m/z 772.6) were detected in the final extracts of porcine liver sample. These endogenous GPs may be the main effectors that cause matrix effects in beta-agonist residue detection in porcine liver.
1. Introduction
Beta-agonists, a group of synthetic phenethanolamine compounds that include clenbuterol, salbutamol, ractopamine, terbutaline, cimaterol, etc. can improve carcass characteristics and promote the growth of animals. However, the illicit use of these compounds in food-producing animals may cause chemical food-poisoning if the meat products obtained from the treated animals are eaten by consumers.1 Therefore, they have been banned as growth promoters of farm animals in several countries.2 Nevertheless, their illegal use in animal husbandry still occurs from time to time.3 Therefore, the monitoring of beta-agonist residues in animal-derived food is required for consumer health.
Because of its high selectivity and sensitivity, liquid chromatography-tandem mass spectrometry (LC-MS/MS) has become a primary analytical technology for multi-residue analysis of veterinary drugs and banned substances in animal-derived food.4,5 However, with the rapid development of this technique and its wide application, a few problems such as matrix effects (MEs) and unavailable qualifier ion for a few compounds have gradually caused concern. ME is a phenomenon in which analyte ionization is suppressed or enhanced due to the interferences co-eluting with the analyte.6 ME can drastically affect the precision, accuracy and linearity of the method and lead to poor and unreliable data in a quantitative assay.7 Both electrospray ionisation (ESI) and atmospheric pressure chemical ionisation (APCI) suffer from ME but ESI has been proven to be more prone to ME than APCI.8 The mechanism of ME is not fully understood but ionization competition between nonvolatile materials and analytes in an ion source is considered to be the most probable cause.9 Therefore, significant attention must be paid to the assessment of ME during the early development of LC-MS/MS methods.10
It has been reported that ME might result from either endogenous components such as lipids (especially phospholipids), proteins, salts, drugs and metabolites,10 which were originally present in samples and retrieved in the final extracts, or exogenous materials such as plastic and polymer residues,11 ion-pairing reagents12 and organic acids,13 which were introduced during sample preparation and analysis. Although both the origins of ME have been widely investigated, endogenous interferences have received more attention in the development of bioanalytical methods.14 Phospholipids have been identified as main matrix effectors and are used as a marker to evaluate ME in biological samples, especially plasma15 in the LC-MS/MS analysis. Bennett et al.16 proposed that the existence of phospholipids in extracts could lead to retention time shifts, elevated baselines, and divergent curves even if they did not co-elute with target analytes. Thus, phospholipids could affect assay performance. Considerable literature is available on the detection, removal and separation of phospholipids in plasma.17 Chambers et al.18 systematically investigated a comprehensive strategy for reducing the ME of drugs in plasma in LC-MS/MS analyses. The combination of polymeric mixed-mode SPE and the appropriate mobile phase pH and UPLC technology provides significant advantages for reducing the matrix effects resulting from plasma matrix components and in improving the ruggedness and sensitivity of bioanalytical methods. However, compared to extensive studies in plasma samples, little attention has been devoted to investigate the influence of phospholipids in edible animal tissues on matrix effects. Moragues et al.19 evaluated matrix effects during the determination of beta-agonists in animal liver and urine using liquid chromatography-ion trap mass spectrometry (more suitable for qualitative analysis). It was suggested that the hexane washing step in solid phase extraction (SPE) could remove phospholipids, fatty acids, etc. responsible, to a great extent, for the ion suppression phenomenon.
The main purpose of this work is to further determine which glycerophospholipids (GPs) are involved in causing matrix effects during the residue analysis of beta-agonists in porcine liver by LC-MS/MS in positive ESI mode. The study focuses on the specific relationship between the amount of GP and the extent of matrix effect of analytes. It is helpful to reduce or compensate matrix effects caused by endogenous phospholipids during the determination of drug residue in porcine liver.
2. Experimental
2.1. Reagents and standards
Standards of salbutamol, terbutaline, cimaterol, fenoterol, ractopamine, clorprenaline, tulobuterol, clenbuterol and penbuterol were provided by Dr Ehrenstorfer (Augsburg, Germany). The standards of lysophosphatidylcholine, LPC (C18:0) and lysophosphatidylcholine, LPC (C16:0) were purchased from Sigma (St Louis, MO, USA), and the standard solutions of phatidylcholine, PC (C16:0/C18:2), phatidylcholine, PC (C16:0/C18:1) and phatidylethanolamine, PE (C18:0/C20:4) in chloroform were provided by Supelco (Bellefonte, PA, USA). Acetonitrile, methanol and formic acid purchased from Fisher Scientific Co. (Pittsburgh, PA, USA) were of HPLC grade. All the other reagents were of analytical grade or higher quality.
Stock solutions of nine beta-agonists were prepared in methanol at a concentration of 1 mg mL−1 and stored at −20 °C. Working solutions were diluted from the stock solutions with 10% methanol in water containing 0.1% formic acid before use in all the experiments except the co-elution study. Standards of LPC C18:0 and LPC C16:0 were first dissolved in a proper amount of chloroform and then diluted to 1 mg mL−1 using methanol (as stock solutions). Working solutions of five GPs were diluted from the stock solutions with 10% methanol in water containing 0.1% formic acid before use.
2.2. Instruments and apparatus
An Agilent 1200 HPLC system (Palo Alto, CA, USA) coupled to an Applied Biosystems API 4000 triple-quadrupole mass spectrometer (Foster City, CA, USA) equipped with an ESI source was employed. A syringe pump (Harvard Apparatus Inc., Holliston, MA, USA) with a 1 mL or 10 mL Hamilton microliter syringe was employed to introduce the solutions into the ESI interface. Universal 32R Centrifuge was purchased from Hettich, Inc. (Tuttlingen, Germany).
2.3. Sample preparation
Porcine liver samples were collected from local markets in China. The liver samples were homogenized and then stored at −20 °C before analysis. A previous analysis was conducted to ensure the absence of target analytes in the porcine liver samples. Samples were treated and analyzed by LC-MS/MS according to a previously developed method.20 In brief, 2 g blank liver sample was extracted with 10 mL of acetonitrile and 1 mL of 10% sodium carbonate solution. The sample was ultrasonicated for about 20 min and centrifuged at 8000 rpm for 10 min at 4 °C. The supernatant was transferred into another tube, and the sediment was then extracted with 5 mL acetonitrile once again. All the supernatants were combined and evaporated in a rotary evaporator until near dryness (about 0.5 mL). Then, the residues were reconstituted in 5 mL of 0.02 mol L−1 ammonium acetate solution (pH = 5.2). The SPE cartridge (a synthesized polymer in acetone), described elsewhere,20 was initially conditioned with 5 mL of methanol followed by 5 mL of water and 5 mL of 0.02 mol L−1 ammonium acetate solution (pH = 5.2), and then the reconstituted extracts were loaded onto the cartridge, which was sequentially washed with 5 mL of water and 5 mL of methanol. Finally, the cartridge was eluted with 5 mL of 4% ammonia in methanol. The eluates were collected, evaporated to dryness under a gentle stream of nitrogen at 50 °C, and the residues reconstituted in 2 mL of 10% methanol in water containing 0.1% formic acid for the subsequent study.
2.4. LC-MS/MS analytical conditions
Chromatographic separation was performed using a Luna C18 column (150 mm × 2.0 mm, 5 μm) purchased from Phenomenex (Torrance, CA, USA). Solvents A and B were 0.1% formic acid in water and acetonitrile, respectively. A gradient elution was used for all the experiments to analyze the nine beta-agonists except for the post-column infusion experiment where isocratic elution (80% B) was adopted. The linear gradient profile consisted of 0–2.0 min: 0–45% B; 2.0–6.0 min: 45% B; 6.0–7.0 min: 45–0% B; 7.0–15 min: 0% B. Flow rate was maintained at 0.25 mL min−1 and column temperature was 35 °C. Injection volume was 5 μL.
The mass analyses were performed using the ESI source in positive ionization mode. Selected reaction monitoring (SRM) experiments were carried out. The operation conditions were as follows: ionspray voltage, 5.0 kV; source temperature, 650 °C; curtain gas, 20 psi; ion source gas 1 and gas 2 were at 60 and 55 psi, respectively. Dwell time was 150 ms for all the nine beta-agonists. Other optimized parameter values and SRM transitions of analytes are given in Table 1 or are published elsewhere.6
Table 1 Selected reaction monitoring settings for MS/MS analysis of nine beta-agonists
| Analyte |
Precursor ions [M + H]+, m/z |
Product ions, m/z |
DPa, V |
CEb, eV |
| DP represents declustering potential. CE represents collision energy. Quantification ions. |
| Ractopamine |
302.2 |
284.2, 164.1c |
54 |
20, 23 |
| Clenbuterol |
277.1 |
258.9, 203.0c |
48 |
19, 23 |
| Salbutamol |
240.0 |
222.0, 148.0c |
60 |
19, 24 |
| Terbutaline |
226.1 |
169.8, 152.1c |
54 |
23, 24 |
| Cimaterol |
220.0 |
143.2, 202.2c |
55 |
21, 16 |
| Fenoterol |
304.2 |
134.8, 107.0c |
54 |
14, 15 |
| Clorprenaline |
214.2 |
196.1, 154.1c |
56 |
17, 18 |
| Tulobuterol |
228.1 |
171.8, 154.1c |
54 |
20, 24 |
| Penbuterol |
292.2 |
201.2, 236.2c |
54 |
30, 25 |
The positive ESI mode was employed to monitor phospholipids. The mass spectrometer acquisition parameters and SRM transitions for the five phospholipids are listed in Table 2. On the basis of literature,17,21 the following characteristic fragment ions were selected as product ions. Specifically, a protonated phosphocholine moiety (product ion, m/z 184) was used to detect PCs, and the positive ion (neutral loss of 141 Da) was used to monitor PE. Other instrument conditions were the same as those used for the nine beta-agonists analysis.
Table 2 Single selected reaction monitoring settings for MS/MS analysis of five phospholipids
| Compounds |
Precursor ion [M + H]+, (m/z) |
Product ions (m/z) |
DPa (V) |
CEb (eV) |
| DP, declustering potential. CE, collision energy. |
| Lysophosphatidylcholine (LPC), C18:0 |
524.4 |
184.2 |
180 |
35 |
| Lysophosphatidylcholine (LPC), C16:0 |
496.2 |
184.2 |
160 |
35 |
| Phosphatidylcholine (PC), C16:0/C18:2 |
758.5 |
184.1 |
156 |
43 |
| Phosphatidylcholine (PC), C16:0/C18:1 |
760.5 |
184.1 |
140 |
45 |
| Phosphatidylethanolamine (PE), C18:0/C20:4 |
768.6 |
627.5 |
138 |
45 |
2.5. Experimental design of the influence of GPs on analyte ionization
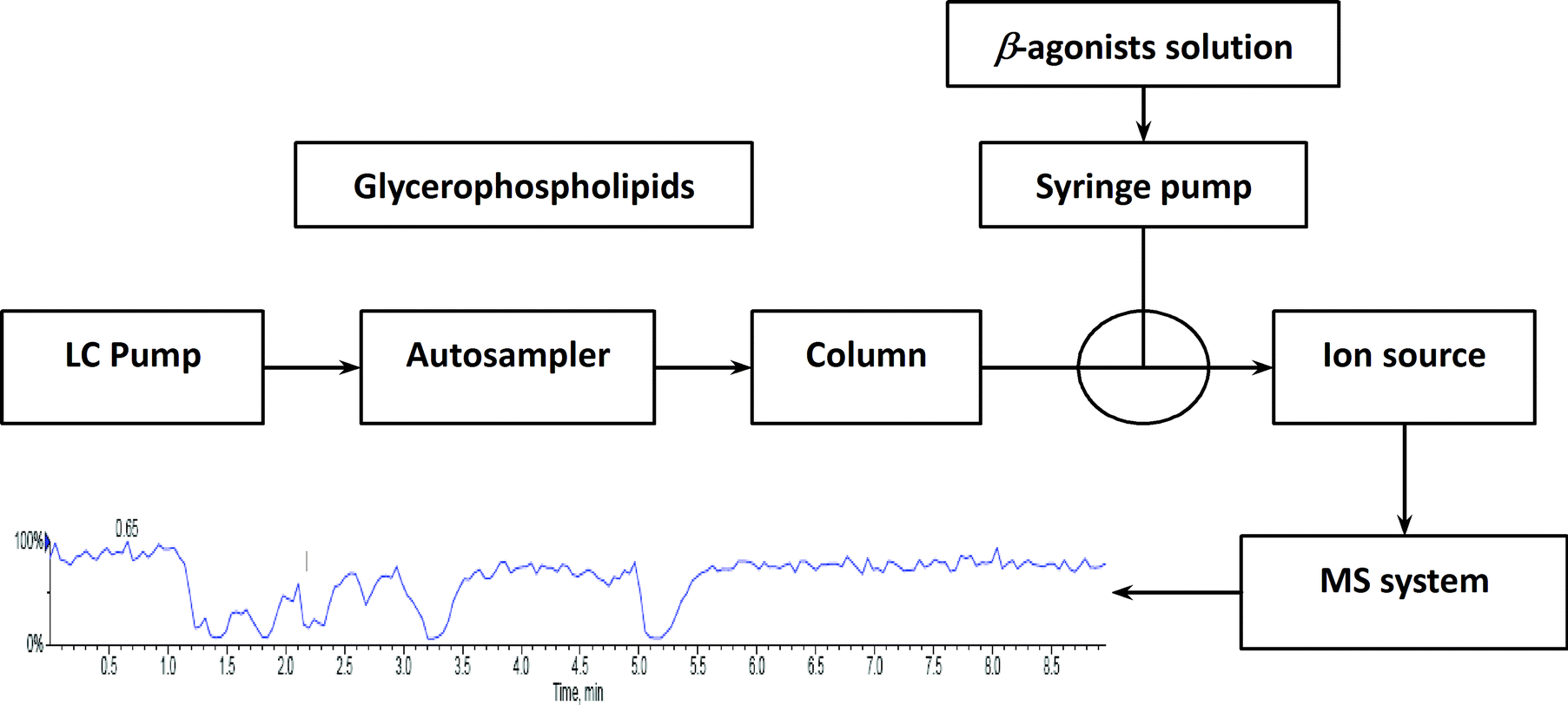
To investigate the influence of phospholipids on analyte ionization, a post-column infusion experiment was carried out. The solution containing nine beta-agonists (each at 2 μg L−1) was constantly infused into the MS system using a syringe pump at a flow rate of 10 μL min−1, while the solution containing five GPs (each at 15.6 mg L−1) was injected in triplicates from the autosampler under isocratic elution (80% B) conditions, as described above. The schematic of the post-column infusion system is shown in Scheme 1. As a comparison, a solution (10% methanol in water containing 0.1% formic acid) was injected from the autosampler as well. Any variation in the signal of the analyte indicated the effect caused by the elution of phospholipids.
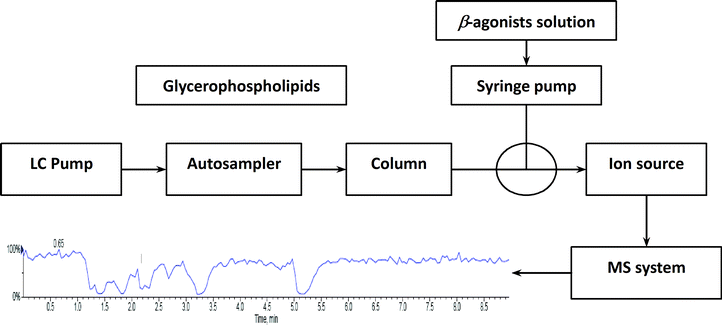 |
| | Scheme 1 Schematic of the post-column infusion system. | |
2.6. Determination of the extent of ion suppression of beta-agonists
First, nine beta-agonists (2 μg L−1) in neat solution (10% methanol in water containing 0.1% formic acid) were analyzed by LC-MS/MS in SRM mode, and peak areas of all the analytes were recorded. Second, the beta-agonists (2 μg L−1) in the solution containing the five GPs at 2.0, 3.9, 7.8, 15.6, 31.3, 62.5 and 125 mg L−1 were prepared and analyzed by LC-MS/MS in SRM mode, respectively. The corresponding peak area of the analyte was recorded. The extent of ion suppression (IS, %) is defined as (100 − B/A × 100),22 where A and B represent the average peak area of the neat standard solution and the standard solution spiked with GPs (n = 5), respectively. Thereafter, the least-squares linear regression line was generated by the IS of each analyte versus the corresponding concentration of GPs and fitted to the equation: y = bx + a, indicating the relationship between the extent of ion suppression of analyte and the concentration of phospholipids. In addition, a linearity test was conducted using statistical analysis by SPSS software package, version 17.0 (SPSS Inc, Chicago, IL, USA). A p-value of <0.01 was considered statistically significant.
2.7. Determination of GPs in the final extracts of porcine liver sample
To find out the presence of GPs in real samples after pretreatment, 20 blank porcine liver tissues from five different markets were extracted with basified acetonitrile and purified by SPE as described above. The final extracts were spiked with a mixture of nine analytes at a concentration level of 2 μg L−1 prior to LC-MS/MS analysis. Besides monitoring the characteristic ion pairs of the phospholipid references, GPs mentioned in the literature,17 such as plasmalogen phosphatidylcholine (PLPC C18 (Plasm)/C18:1, m/z 772.6/184.1) and lysophosphatidylethanolamine (LPE C16:0, m/z 454.3/313.3), were also monitored during the LC-MS/MS analysis.
3. Results and discussion
Post-extraction addition and post-column infusion are the two main approaches used to evaluate ME. In post-extraction addition, the value of ME is evaluated as the ratio of analyte response in blank sample extracts to the response of the same concentration in a neat solution.8 This provides information regarding ME at the time of analyte elution. Post-column infusion is carried out by monitoring the response of an analyte constantly infused by an infusion pump, and then injection of blank sample extracts using the LC system under the desired chromatographic conditions.23 By this technique, chromatographic regions where an analyte most likely suffers from ME can be identified. If the main sources that give rise to ME are identified, we can remove them more accurately. Thus, matrix effects will be overcomed or diminished.
3.1. Influence of GPs on analyte ionization
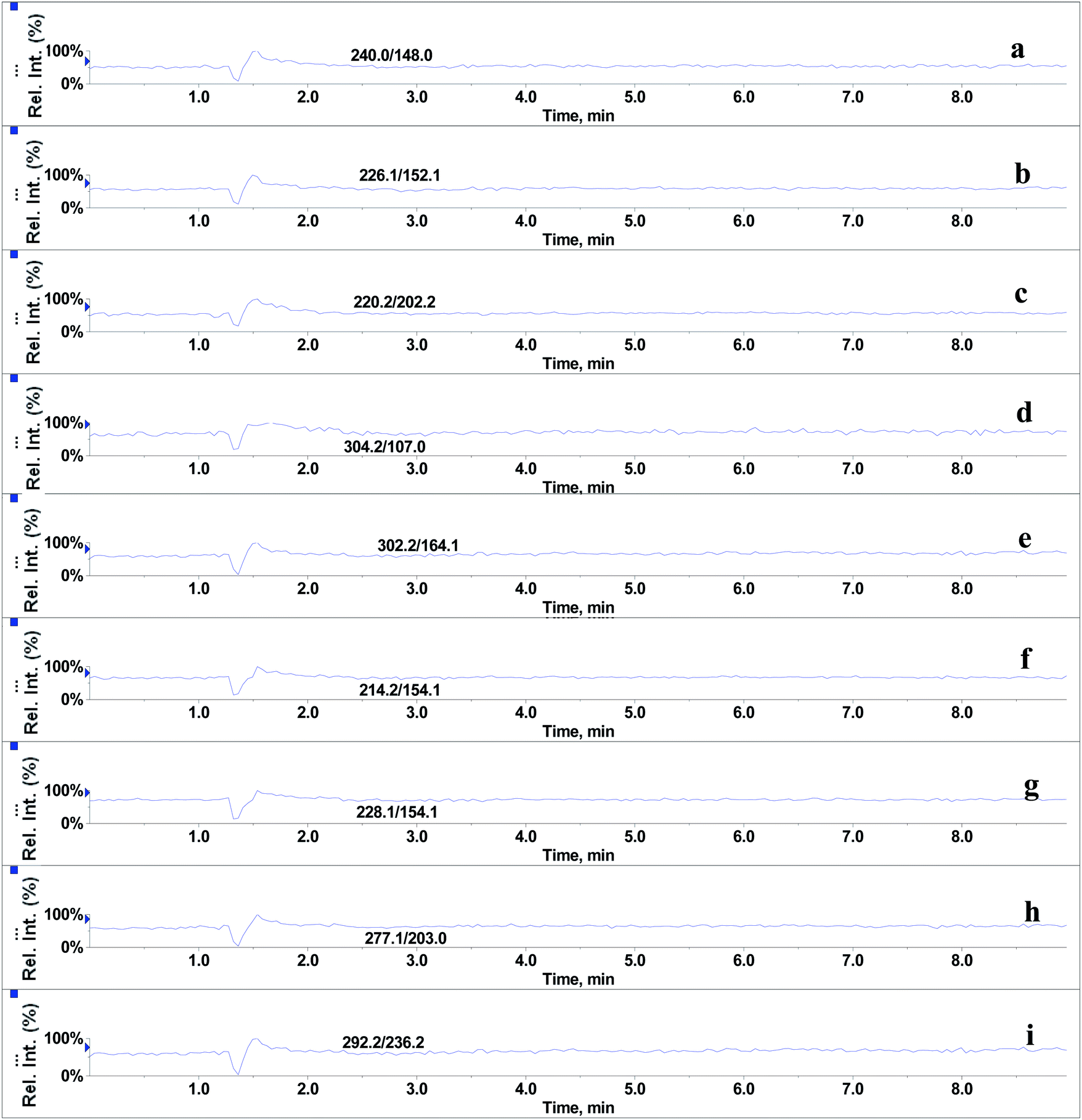
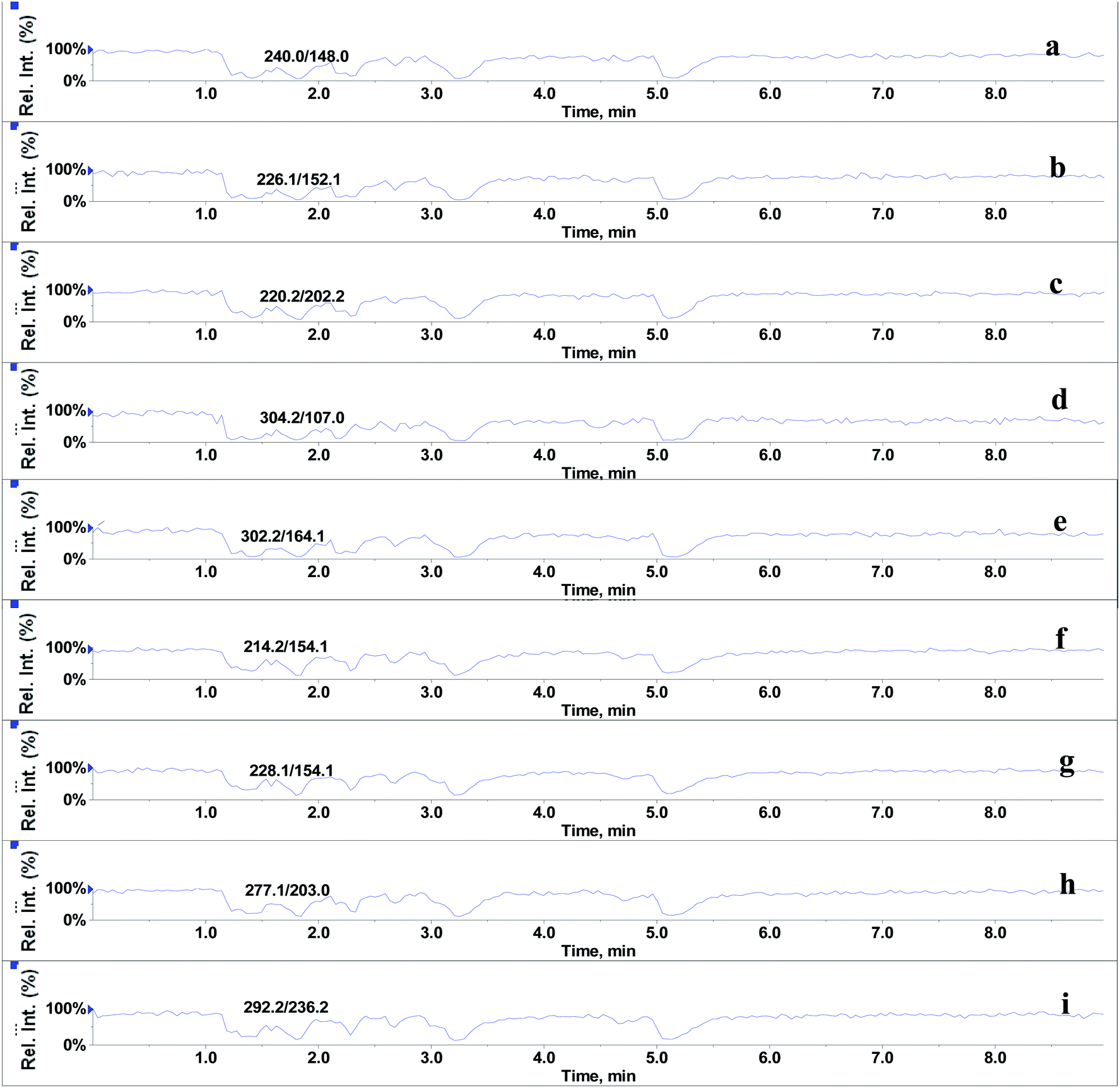
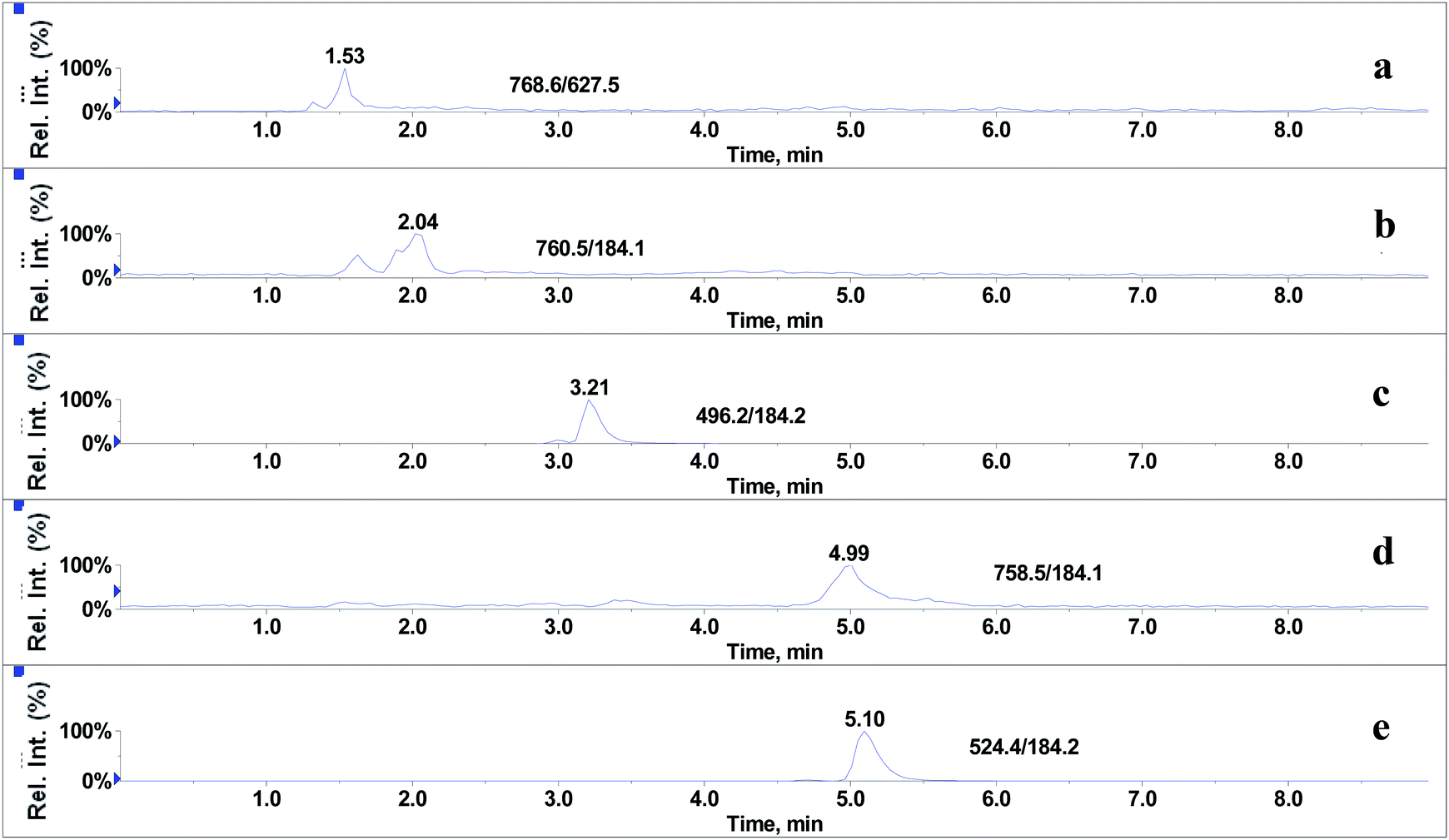
In the absence of any interferences eluting from the analytical column, the post-column infusion of an analyte should obtain a constant background signal in the SRM channel. Any variation in the constant signal profile of the analyte indicates the presence of matrix effects. To investigate the effects of phospholipids on analyte ionization, the neat solvent and mixed GPs solution of LPC (C18:0), LPC (C16:0), PC (C16:0/C18:2), PC (C16:0/C18:1) and PE (C18:0/C20:4) were injected via an autosampler, while analytes were being constantly infused from post-column. The results showed that for the neat solvent (10% methanol in water containing 0.1% formic acid) injected pre-column, steady signal profiles for nine beta-agonists were observed, except for a dip at about 1.3 min due to injection (see Fig. 1). On the contrary, the results following the injection of the GP solution presented several significant signal dips (see Fig. 2). Therefore, it was suggested that ion suppression of the analyte was caused by the presence of GPs. Representative chromatograms of five phospholipids are shown in Fig. 3. It was observed that one peak was eluted for PE (C18:0/C20:4) (Fig. 3a) and PC (C16:0/C18:1) (Fig. 3b), which was why more than two signal dips were produced at the elution time windows of two phospholipids (see Fig. 2). Furthermore, the peaks of PE (C18:0/C20:4) and PC (C16:0/C18:2) (Fig. 3d) were partially overlapping with those of PC (C16:0/C18:1) and LPC (C18:0) (Fig. 3e), which led to an increase of the signal suppression at time windows of 5 min (see Fig. 2).
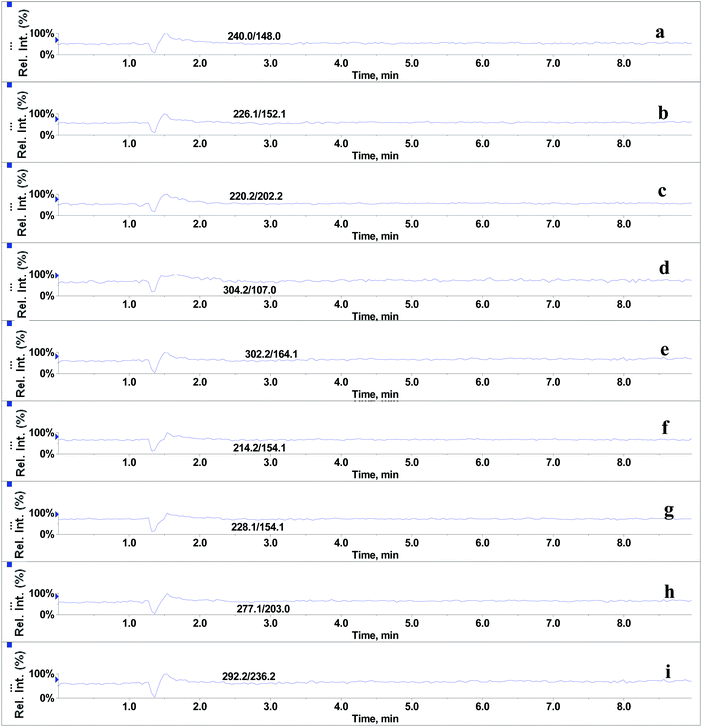 |
| | Fig. 1 Post-column infusion spectra of beta-agonists (2 μg L−1) after injection of solvent (10% methanol in water containing 0.1% formic acid): (a) salbutamol, (b) terbutaline, (c) cimaterol, (d) fenoterol, (e) clorprenaline, (f) ractopamine, (g) tulobuterol, (h) clenbuterol and (i) penbuterol. | |
 |
| | Fig. 2 Post-column infusion spectra of beta-agonists (2 μg L−1) after injection of glycerophospholipids standards mixture (15.6 mg L−1). (a–i) identification as in Fig. 1. | |
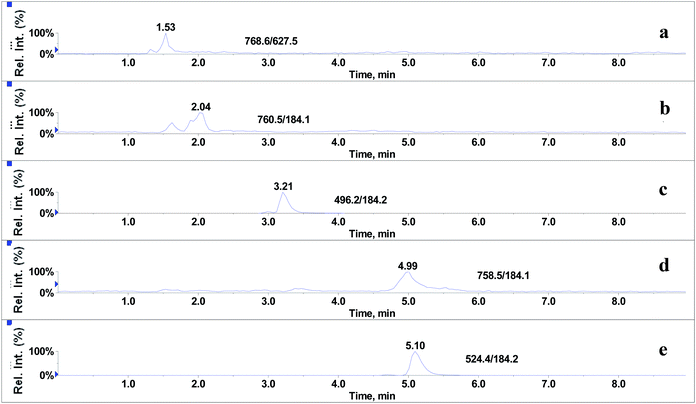 |
| | Fig. 3 Representative chromatograms of glycerophospholipids standards: (a) PE (C18:0/C20:4), (b) PC (C16:0/C18:1), (c) LPC (C16:0), (d) PC (C16:0/C18:2) and (e) LPC (C18:0). | |
3.2. Relationship between extent of IS and amount of GPs
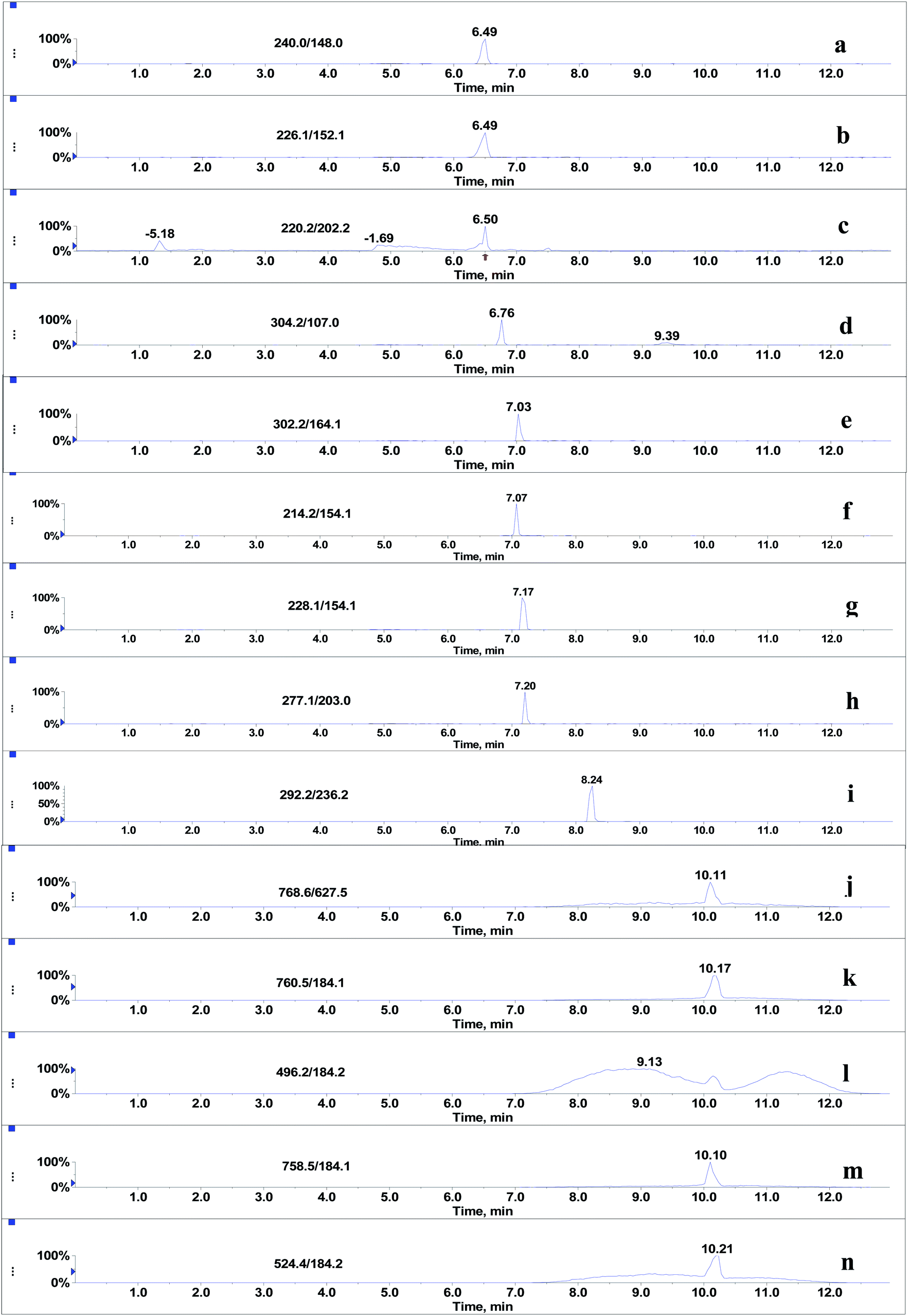
Since the post-column infusion experiment demonstrated that GPs caused significant ion suppression for the nine beta-agonists, the co-elution of GPs and target analytes was further detected. First, the mixed standards solution of nine beta-agonists and five GPs was prepared, and the most intensive SRM transition of each compound was monitored during the LC-MS/MS analysis. The results showed that the retention time windows (peak widths) of five GPs ranged from 7.0 to 13 min. Most target analytes including fenoterol, ractopamine, clorprenaline, tulobuterol, clenbuterol and penbuterol were also eluted during these periods and were not well separated from GP interferences. Typical extracted ion mass chromatograms of the co-eluates (nine beta-agonists and five GPs) are shown in Fig. 4. Three beta-agonists including salbutamol, terbutaline and cimaterol were eluted at 6.5 min.
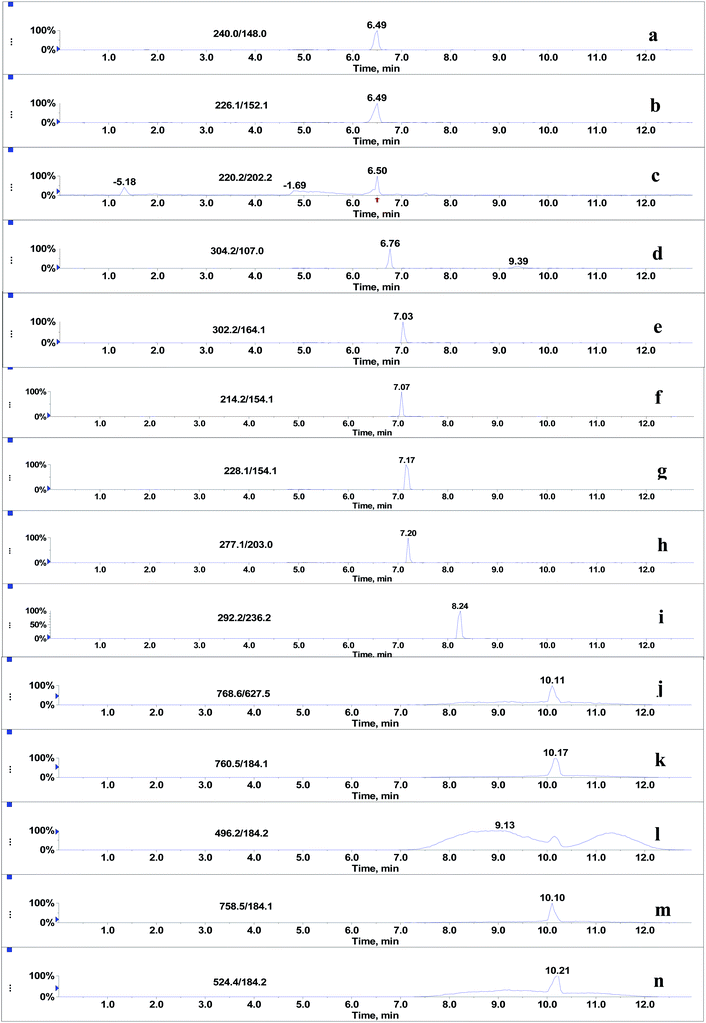 |
| | Fig. 4 Co-elution of nine beta-agonists (2 μg L−1) and five glycerophospholipids standards (125 mg L−1): (a) salbutamol, (b) terbutaline, (c) cimaterol, (d) fenoterol, (e) clorprenaline, (f) ractopamine, (g) tulobuterol, (h) clenbuterol, (i) penbuterol, (j) PE (C18:0/C20:4), (k) PC (C16:0/C18:1), (l) LPC (C16:0), (m) PC (C16:0/C18:2) and (n) LPC (C18:0). | |
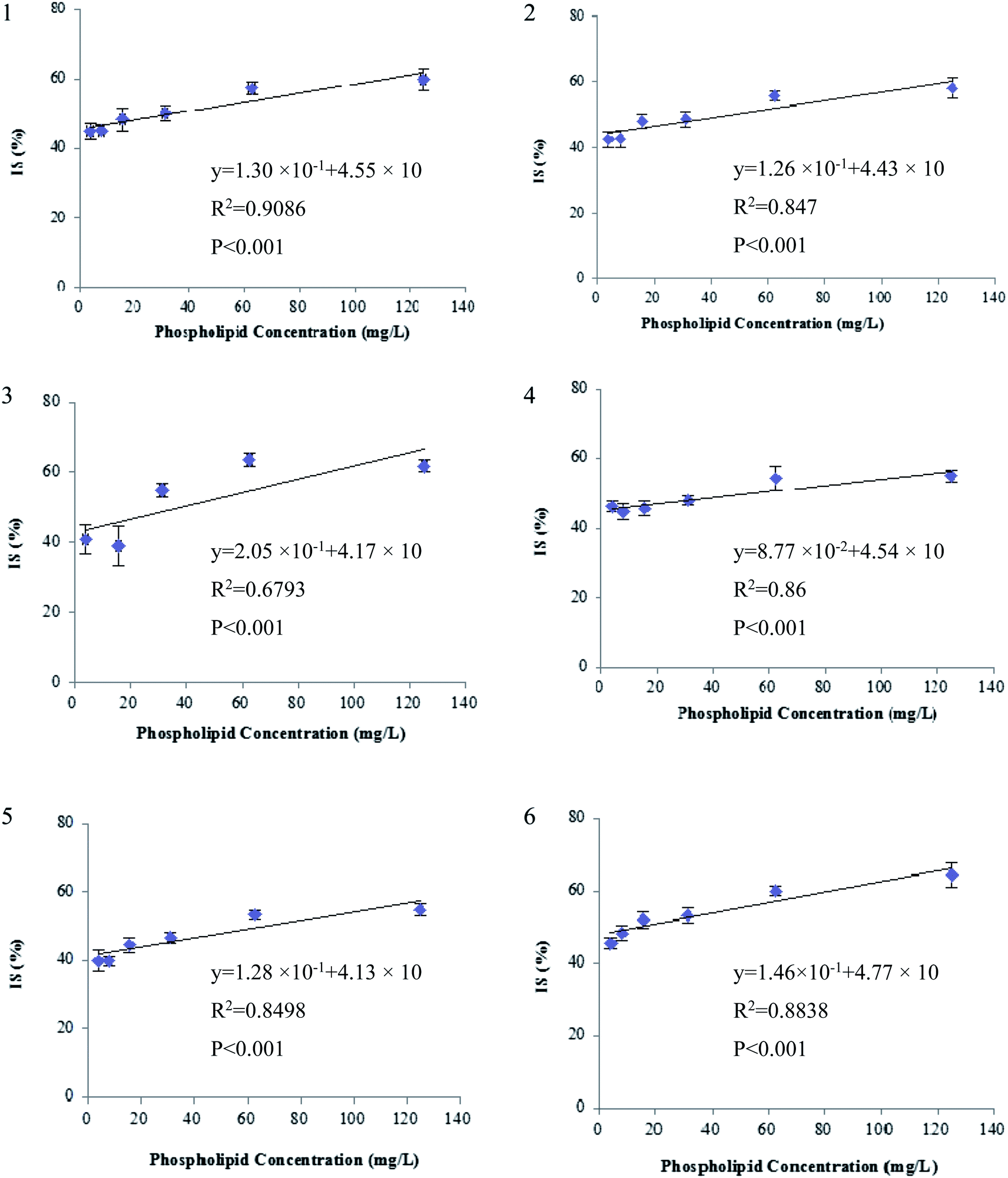
Second, the extent of ion suppression was determined under different concentrations of GPs (15.6 mg L−1, which is the maximal mixed concentration obtained by diluting the purchased GP solutions), and IS of analytes versus the concentration of GPs was fitted with least-squares linear regression. The results show that a positive linear correlation is observed between the GPs and each of the six beta-agonists including fenoterol, ractopamine, clorprenaline, tulobuterol, clenbuterol and penbuterol, which were co-eluted with GPs. The regression curves and correlation coefficients are shown in Fig. 5. The R2 values of the six beta-agonists, except for ractopamine (0.68), were more than 0.85. In addition, the results of a linearity test of the curves showed that p-values were all <0.001 for R2 of the six analytes, indicating that the linear correlation was statistically significant. Thus, for these target analytes the extent of IS increased linearly with the increase of GPs in porcine liver sample. Therefore, we could deduce that GPs played an important role in causing the matrix effects of beta-agonists in LC-MS/MS analysis.
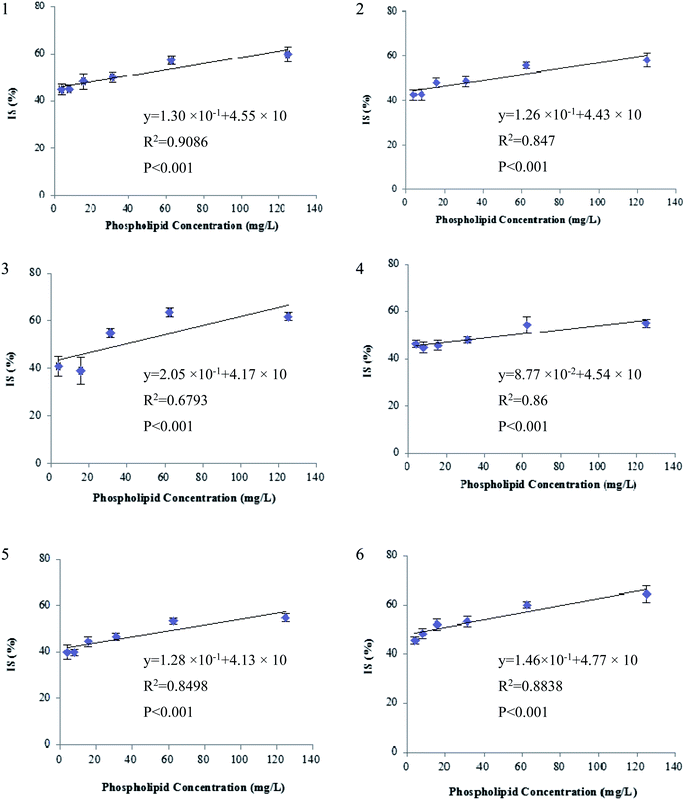 |
| | Fig. 5 Correlation between ion suppression (IS) of beta-agonists and phospholipids concentration (n = 6): (a) fenoterol, (b) clorprenaline, (c) ractopamine, (d) tulobuterol, (e) clenbuterol and (f) penbuterol. IS (%) = 100 − B/A × 100, where A and B represent peak area of analyte in pure solvent and in solution spiked with different amounts of phospholipids, respectively. The regression curves and correlation coefficients were obtained using EXCEL, version 2003, and p-value was obtained by statistical software package SPSS, version 17.0. | |
3.3. GPs in the final extracts of liver samples
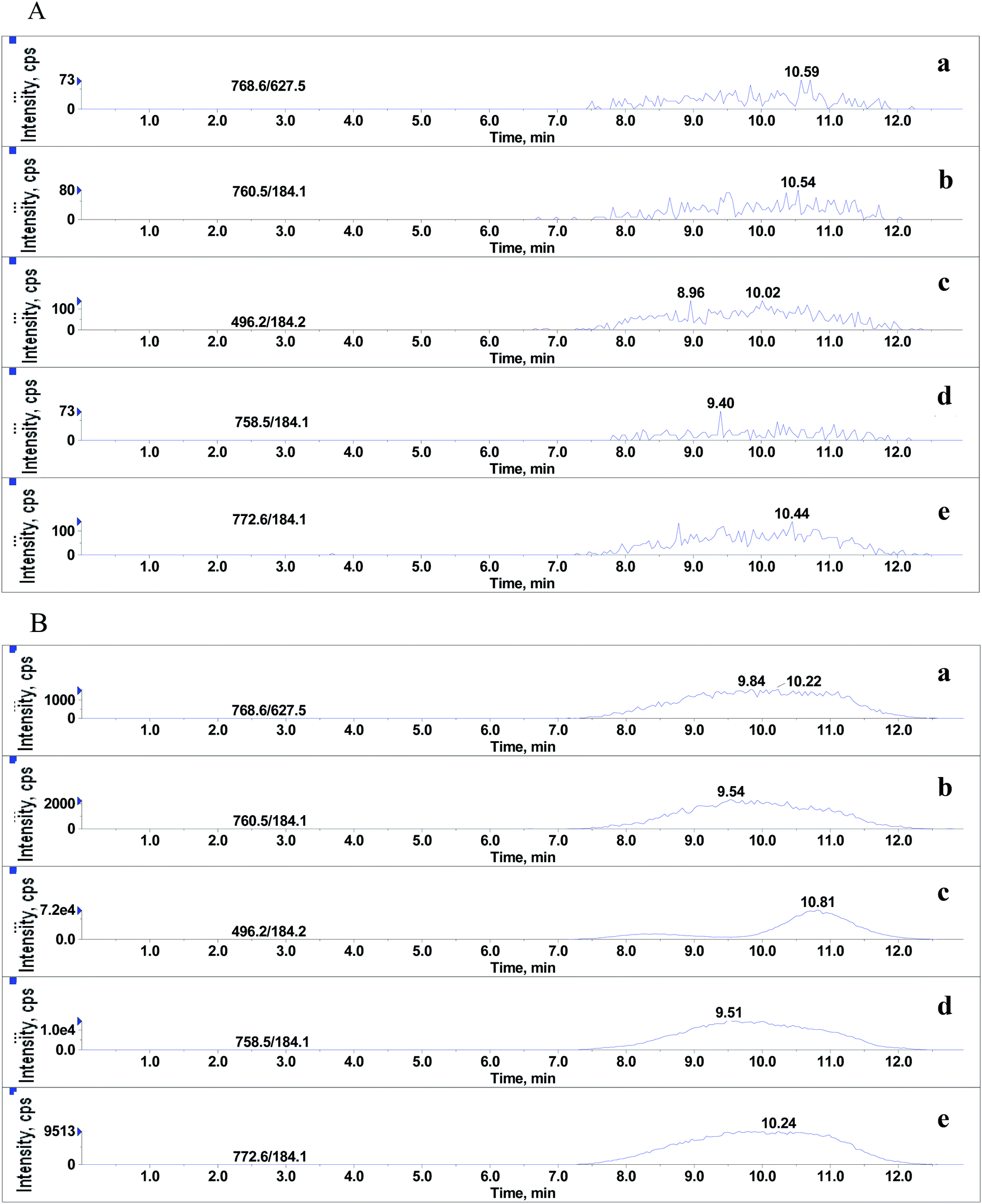
Although it was proved that GPs had significant effects on MEs of the beta-agonists, whether they still existed in the final extracts of real sample was yet to be identified. After extraction with basified acetonitrile followed by purification using weak cation exchange SPE,20 the sample extracts were injected into LC-MS/MS system under the optimized conditions for beta-agonists. As shown in Fig. 6, significant amounts of endogenous GPs were detected in porcine liver samples. The LPC (m/z 496.2), PC (m/z 758.5), PC (m/z 760.5), PE (m/z 768.6) and PLPC (m/z 772.6) presented large and wide peaks, and their peak heights ranged from 1.6 × 103 to 7.2 × 104 and peak widths from 5.4 min to 5.7 min. The results suggested that the GPs, which remained in the extracts of porcine liver tissues had significant effect on the MEs of beta-agonists. In our experiments, hexane was also used to remove phospholipids before and after SPE. The results indicate that hexane can remove phospholipids to some extent but the difference between the steps using hexane or not is not significant (p > 0.05), and SPE using a weak cation polymer synthesized in our laboratory might significantly remove phospholipids in porcine liver samples (p < 0.05). SPE is better than hexane to remove phospholipids, which is consistent with the findings reported by Chambers18et al. Complete removal of phospholipids from animal tissue samples is difficult. Various efficient strategies have been used to reduce or compensate matrix effects such as optimizing sample preparation, manipulating chromatographic parameters and using a stable isotope internal standard; however, in some cases, the above-mentioned strategies cannot be performed. The strategy of spiking appropriate GPs to standard solution (naming standard addition method) for compensating matrix effects in complex biological samples is considerable in LC-MS/MS analysis. The matrix-matched calibration curve should be adopted for reliable quantification.
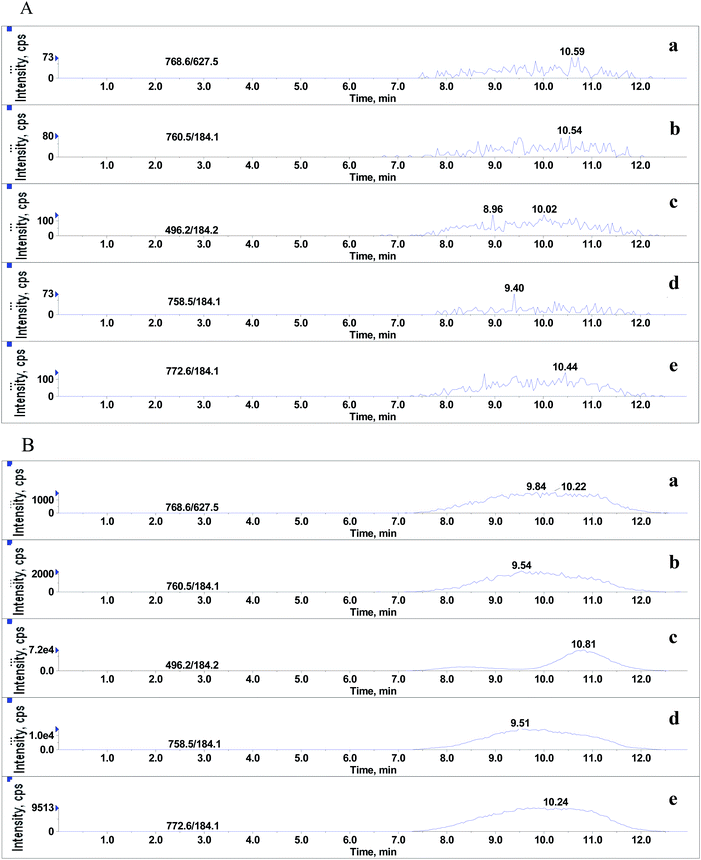 |
| | Fig. 6 Typical SRM chromatograms of 5 phospholipids. (A) and (B) represent final extracts of porcine liver sample and 10% methanol in water containing 0.1% formic acid, respectively: (a) PE (C18:0/C20:4), (b) PC (C16:0/C18:1), (c) LPC (C16:0), (d) PC (C16:0/C18:2) and (e) C18:0 (Plasm)/C18:1 PLPC. | |
4. Conclusions
Glycerophospholipids might cause significant ion suppression effects on the signals of nine beta-agonists including salbutamol, terbutaline, cimaterol, fenoterol, ractopamine, clorprenaline, tulobuterol, clenbuterol and penbuterol. A positive linear correlation (p < 0.01) was observed between the extent of ion suppression of five beta-agonists and the amount of phospholipids with correlation coefficients greater than 0.85. Considerable amounts of phospholipids such as LPC (m/z 496.2), PC (m/z 758.5), PC (m/z 760.5), PE (m/z 768.6) and PLPC (m/z 772.6) were detected in the final extracts of porcine liver samples. Glycerophospholipids were suggested to be used as general substances to compensate matrix effects in complex biological samples with LC-MS/MS analysis.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 31072166).
References
- J. F. Martinez-Navarro, Lancet, 1990, 336, 1311 CrossRef CAS PubMed
 .
. - Council Directive 96/22/EC of 29 April 1996 concerning the prohibition on the use in stock farming of certain substances having a hormonal or thyrostatic action and of β-agonists, and repealing Directives 81/602/EEC, 88/146/EEC and 88/299/EEC, Off. J. Eur. Communities: Legis., 1996, 10–31.
- S. Fan, H. Miao, Y. Zhao, H. Chen and Y. Wu, J. Agric. Food Chem., 2012, 60, 1898–1905 Search PubMed .
- B. Shao, X. Jia, J. Zhang, J. Meng, Y. Wu, H. Duan and X. Tu, Food Chem., 2009, 114, 1115–1121 CrossRef CAS .
- C. Li, Y. L. Wu, T. Yang, Y. Zhang and W. G. Huang-Fu, J. Chromatogr. A, 2010, 1217, 7873–7877 CrossRef CAS PubMed .
- L. Q. Wang, Z. L. Zeng, Y. J. Su, G. K. Zhang, X. L. Zhong, Z. P. Liang and L. M. He, J. Agric. Food Chem., 2012, 60, 6359–6363 CrossRef CAS PubMed .
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 1998, 70, 882–889 CrossRef CAS PubMed .
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 75, 3019–3030 CrossRef CAS PubMed .
- R. King, R. Bonfiglio, C. Fernandez-Metzler, C. Miller-Stein and T. Olah, J. Am. Soc. Mass Spectrom., 2000, 11, 942–950 CrossRef CAS PubMed .
- J. P. Antignac, K. De Wasch, F. Monteau, H. De Brabander, F. O. Andre and B. Le Bizec, Anal. Chim. Acta, 2005, 529, 129–136 CrossRef CAS .
- H. Mei, Y. Hsieh, C. Nardo, X. Xu, S. Wang, K. Ng and W. A. Korfmacher, Rapid Commun. Mass Spectrom., 1999, 13, 1175–1185 CrossRef .
- C. R. Mallet, Z. Lu and J. R. Mazzeo, Rapid Commun. Mass Spectrom., 2004, 18, 49–58 Search PubMed .
- J. Eshraghi and S. K. Chowdhury, Anal. Chem., 1993, 65, 3528–3533 Search PubMed .
- S. Silvester and L. Smith, Bioanalysis, 2012, 4, 879–895 CrossRef CAS PubMed .
- O. A. Ismaiel, M. S. Halquist, M. Y. Elmamly, A. Shalaby and H. Thomas Karnes, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 875, 333–343 CrossRef CAS PubMed .
- P. Bennett and H. Liang, in ASMS Conference, Nashville, Tennesee, 2004 Search PubMed .
- Y. Q. Xia and M. Jemal, Rapid Commun. Mass Spectrom., 2009, 23, 2125–2138 CrossRef CAS PubMed .
- E. Chambers, D. M. Wagrowski-Diehl, Z. L. Lu and J. R. Mazzeo, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 852, 22–34 CrossRef CAS PubMed .
- F. Moragues and C. Igualada, Anal. Chim. Acta, 2009, 637, 193–195 CrossRef CAS PubMed .
- L. Wang, Z. Zeng, X. Wang, J. Yang, Z. Chen and L. He, J. Sep. Sci., 2013, 36, 1843–1852 CrossRef CAS PubMed .
- M. Pulfer and R. C. Murphy, Mass Spectrom. Rev., 2003, 22, 332–364 CrossRef CAS .
- D. L. Buhrman, P. I. Price and P. J. Rudewicz, J. Am. Soc. Mass Spectrom., 1996, 7, 1099–1105 CrossRef CAS PubMed .
- R. Bonfiglio, R. C. King, T. V. Olah and K. Merkle, Rapid Commun. Mass Spectrom., 1999, 13, 1175–1185 CrossRef CAS .
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.