
Nanoporous morphology control of polyethylene membranes by block copolymer blends†
Abstract
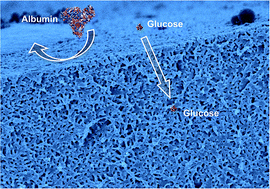
Nanoporous polyethylene (PE) membranes prepared by etching treatment for diblock copolymers composed of PE and polystyrene (PS) are applicable for biotechnological interfaces of various devices. The conventional control of pore size is limited by block compositions or etching conditions. In this study, an increase in the removable PS component was enabled by blending the above base copolymer (PE-b-PS) with PS homopolymer or another block copolymer containing a greater PS content. A series of films of these blends were isothermally crystallized under the optimum conditions, and were mildly etched to prepare nanoporous membranes. Therefore, excellent robustness was achieved in the resultant nanoporous membranes, which is beneficial for wider applications. A desirable combination of precise pore dimensions and large pore volume was achieved by blending the PS homopolymer with lower molecular weight, compared to a nanoporous membrane prepared from pure base copolymer, which is also confirmed by molecular permeation tests.
 Please wait while we load your content...
Please wait while we load your content...

