
Ruthenium-catalyzed cross-metathesis with electron-rich phenyl vinyl sulfide enables access to 2,3-dideoxy-d-ribopyranose ring system donors†
Abstract
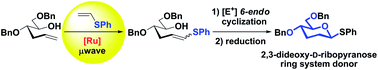
2,3-Dideoxy-D-ribopyranose units are important ring systems found in nature. Herein, we develop a metal-mediated strategy to form this important scaffold featuring a cross-metathesis reaction of the corresponding sugar-derived hydroxyalkene with electron-rich phenyl vinyl sulfide using commercially available ruthenium-catalysts under microwave irradiation as a key step. The final 2,3-dideoxyhexopyranose ring is generated in a single step upon 6-endo electrophilic cyclization.
 Please wait while we load your content...
Please wait while we load your content...

