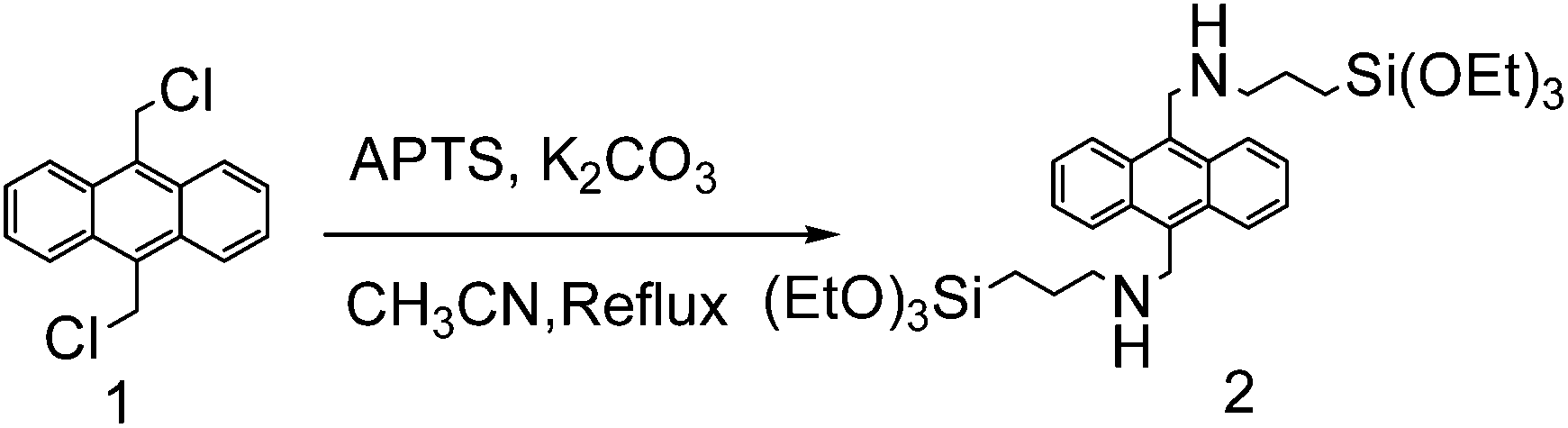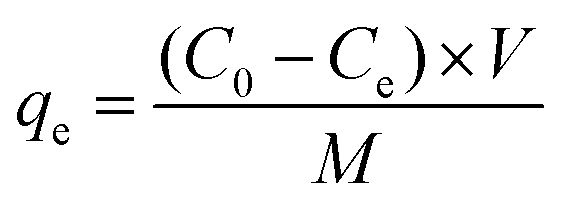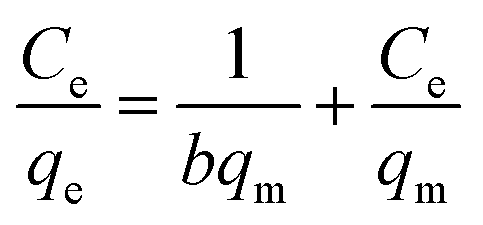DOI:
10.1039/C4RA01599A
(Paper)
RSC Adv., 2014,
4, 18660-18667
Self-assembly of magnetically recoverable ratiometric Cu2+ fluorescent sensor and adsorbent
Received
24th February 2014
, Accepted 26th March 2014
First published on 1st April 2014
Abstract
A mesoporous shell containing a fluorescence resonance energy transfer (FRET) type of ratiometric Cu2+ fluorescent sensor was coated around a cetyltrimethylammonium bromide (CTAB) stabilized Fe3O4@SiO2 core. All of the units comprising the sensor composite, including the Cu2+ ligand, signal reference and reporter units, are independent of each other and without a direct covalent linkage between them, and are site-selectively self-assembled into the pore framework or channel of the mesoporous silica matrix through the electrostatic interaction between CTAB porogen and silicates. The coordination of Cu2+ and its ligand leads to the variation of fluorescence energy transfer efficiency between neighboring FRET pairs benefiting from the nanosized pore system of the mesoporous matrix, which finally results in the ratiometric detection of Cu2+. Investigation of Cu2+ adsorption performance indicated rapid removal efficiency, with a maximum adsorption capacity of 17.86 mg g−1. Fe3O4 nanoparticles were introduced as a core to make the sensor system magnetically recyclable after sensing and adsorption. Finally, the disassembly and reassembly of the Cu2+ sensor were achieved by extracting and reintroducing the units in CTAB micelles, making the sensing system reproducible.
Introduction
Surfactant chemistry has regained its youth since the rise of nanomaterial science, attributed to its indispensable role in stabilization of nanometer-sized metal or metal oxide and excellent performance on assembly of an all-in-one multifunctional system. A typical example is a core–shell type of nanoparticle composed of a metal or metal oxide core and mesoporous shell, which is assembled from a surfactant-stabilized core and a surfactant-templated mesoporous shell.1–4 In this system, the surfactant perfectly plays the role of bridging two or more different functional units, which actually can be freely alternated according to the application purpose. Up to now, using core–shell nanoparticles (NPs) with a mesoporous silica shell as examples, a library of nano-assembly NPs has been fabricated – for example, Au@mSiO2, Fe3O4@mSiO2, QD@mSiO2, etc.5–7 In addition to its recent application in nanoscience, a surfactant is traditionally recognized by its excellent ability to decrease surface tension and solubilize hydrophobic organics.8 For example, by adopting a system composed of surfactant and hydrophobic metal ion ligands, cloud point extraction technology is often used for the concentration and separation of metal ions.9–11
Here, with the help of the clever combination of old and new surfactant science, we develop a novel one-pot self-assembly method for the fabrication of a magnetically recyclable ratiometric Cu2+ fluorescent sensor and adsorbent based on mesoporous silica. Fluorescent sensors are well-known for their advantages such as real-time, in site, convenience and accuracy, and have been elaborately designed and synthesized for the detection of ions and molecules in the environmental and biological fields.12–19 However, they are obstructed from practical applications by disadvantages such as low sensitivity, complicated procedures for molecular fabrication, and poor hydrophilicity.20–26 Much effort has been devoted to simplifying the synthesis procedure and improving the sensing performance. For example, detection sensitivity can be improved by combining an organic sensor with nanoparticles, owing to the improved contact efficiency between sensor and target.27–35
However, the fabrication process, especially for ratiometric sensors, is still complicated and requires multiple steps to covalently link different functional groups. It is still desirable to establish a simpler and more convenient method for sensor synthesis. We previously reported a pH ratiometric sensor using mesoporous silica as matrix and two commercially available fluorophores as signal reference and reporter units, where the detection sensitivity was greatly improved.36 In this paper, we have further developed a simple way for fabrication of a multifunctional system assembling sensing, adsorption and magnetic recovery in one NP through artfully combining the old and new chemistry of surfactants. We adopted a cetyltrimethylammonium bromide (CTAB) stabilized Fe3O4@SiO2 NP as the core and coated a fluorescent mesoporous shell around it, which was self-assembled through the electrostatic force between cationic CTAB containing a Cu2+ ligand and fluorescein derivative and anionic silicates encapsulated with anthracene. All of the units are just simply mixed in the synthesis system, and then ultimately select the sites they tend to occupy in an autonomous way, which is actually driven by multiple forces including electrostatic, “like-dissolves-like”, and silicate condensation. The basic purpose of this paper is to simplify the troublesome fabrication process of the traditional fluorescent sensor and improve its detection performance by multifunctionalization with the aid of versatile surfactant chemistry, and then ultimately present a good example and means for fabrication and assembly of novel materials.
Experimental
Materials
FITC and bischloromethylanthracene were acquired from Amresco. (Boc)2O, CF3OOH, 1-bromodecane and glycine ethyl ester hydrochloride were supplied by Shanghai Kayon Biological Technology Co. Ltd. CTAB, absolute ethanol, TEOS and ammonium hydroxide were purchased from Shanghai Sinopharm Chemical Reagent Co. Ltd., China. APTS was supplied by Shanghai Yaohua Chemical Plant. Distilled deionized water was used for the preparation of all aqueous solutions. All starting materials and solvents were used as received without further purification.
Preparation of bis-silylated anthracene
To a solution of 1 (0.548 g, 2 mmol) and K2CO3 (1.09 g, 8 mmol) in anhydrous CH3CN (40 mL) solution, APTS (0.96 mL, 4 mmol) was added dropwise. The mixture was then heated to reflux for 48 h under a N2 atmosphere. After the reaction was completed, the solvent was removed under reduced pressure. The crude product was then purified by chromatography on a silica gel column (ethyl acetate) to give the target product as a pale yellow liquid (192 mg, 15% yield); 1H NMR (400 MHz CDCl3) δ 0.68 (t, 4H, CH2), 1.19 (t, 18H, CH3), 1.68–1.74 (m, 4H, CH2), 2.54 (s, 2H, NH), 2.88 (t, 4H, CH2), 3.79 (q, 12H, CH2), 4.71 (s, 4H, CH2), 7.52 (t, 4H, ArH), 8.38 (d, 4H, ArH); 13C NMR (100 MHz CDCl3) δ: 8.05, 18.32, 23.39, 45.89, 53.43, 58.38, 124.94, 125.60, 126.46, 126.89, 127.59, 130.13, 132.20.
Preparation of Cu2+ ligand
Preparation of intermediate 2. Glycine ethyl ester hydrochloride (8.84 g, 40 mmol) was added to a solution of 1-bromodecane (8.84 g, 40 mmol) and triethylamine (2.75 mL, 19.6 mmol) in 50 mL of ethanol. The reaction mixture was refluxed for 6 h. After this time, the mixture was cooled to room temperature, 150 mL of CHCl3 was added, and the organic phase was extracted with 0.5 M HCl (3 × 150 mL), a 10% NaHCO3 solution (3 × 150 mL), and water (3 × 150 mL). The organic phase was dried (Na2SO4), and the solvent was evaporated. The crude product was purified by chromatography on a silica gel column (CH2Cl2) to give the target product as a yellow liquid (2.4 g, 25% yield); 1H NMR (400 MHz CDCl3), δ: 0.88 (t, 3H, J = 3.2 Hz), 1.27 (t, 17H, J = 11.2 Hz), 1.49 (d, 2H, J = 5.6 Hz), 1.67 (s, 1H), 2.59 (m, 2H, J = 4 Hz), 3.39 (d, 2H, J = 6.4 Hz), 4.19 (m, 2H, J = 3.6 Hz); 13C NMR (100 MHz, CDCl3), δ: 14.13, 22.58, 27.16, 29.45, 29.99, 31.82, 49.58, 50.95, 60.52, 172.46.
Preparation of intermediate 3. A solution of di-tert-butyl dicarbonate (1.89 g, 1.05 mmol) in 10 mL of CHCl3 was added dropwise to a solution of the above N-alkylamino ester (2.2 g, 1 mmol) in 50 mL of CHCl3. The reaction mixture was stirred at room temperature overnight, and then extracted with a 5% solution of Na2CO3. The organic phase was dried (Na2SO4), and the solvent was evaporated to yield the proper N-tert-butoxycarbonyl-N-alkylglycine ethyl ester as a yellow oil (2.99 g, 98% yield). 1H NMR (400 MHz CDCl3), δ: 0.88 (t, 3H, J = 3 Hz), 1.27 (t, 17H, J = 11.2 Hz), 1.43 (t, 4H, J = 2 Hz), 1.48 (d, 4H, J = 2 Hz), 1.53 (t, 3H, J = 2.8 Hz), 3.27 (m, 2H, J = 6.8 Hz); 13C NMR (100 MHz CDCl3), δ: 14.04, 22.62, 26.73, 27.35, 28.26, 29.50, 31.84, 48.34, 48.69, 49.32, 60.84, 79.84, 170.14.
Preparation of intermediate 4. A 2 M solution of NaOH (10 mL, 20 mmol) was added to a solution of this Boc-protected derivative (0.72 g, 2 mmol) in MeOH (10 mL). The reaction mixture was stirred for 3 h at room temperature, and then the pH of the solution was adjusted to about 7 with 1 M HCl, and the product was collected by filtration as a white solid (0.45 g, 99% yield). 1H NMR (400 MHz CDCl3), δ: 0.88 (t, 3H, J = 6.8 Hz), 1.25 (s, 14H), 1.42 (s, 11H), 3.18 (t, 2H, J = 6.8 Hz), 3.61 (s, 2H); 13C NMR (100 MHz CDCl3), δ: 14.09, 22.68, 26.91, 28.41, 29.60, 31.94, 48.55, 51.46, 79.38, 156.97, 177.19.
Preparation of intermediate 5. A solution of the above N-tert-butoxycarbonyl-N-alkylglycine (0.66 g, 2 mmol) in CH2Cl2 was added to hydroxybenzotriazole (HOBt, 284 mg, 2.1 mmol), N-(3-dimethylaminopropyl)-N-ethylcarbodiimide (EDC, 402 mg, 2.1 mmol), glycine ethyl ester hydrochloride (280 mg, 2 mmol) and triethylamine (280 μL, 2 mmol) in anhydrous CH2Cl2 (20 mL). The reaction mixture was stirred at room temperature overnight. After that, 30 mL of ethyl acetate was added, and the mixture was extracted with a 0.5 M citric acid solution (2 × 30 mL), a 5% NaHCO3 solution (2 × 30 mL), and water (2 × 30 mL). The organic phase was dried with Na2SO4, and the solvent was removed to yield the proper protected N-alkyl dipeptide as a yellow oil (0.82 g, 99%) and without further purification for the next step; 1H NMR (400 MHz CDCl3), δ: 0.88 (t, 3H, J = 6.8 Hz), 1.29 (t, 17H, J = 7 Hz), 1.47 (s, 8H), 1.53 (t, 3H, J = 7 Hz), 3.28 (t, 2H, J = 7.4 Hz), 3.42 (s, 2H), 3.90 (s, 2H), 4.20 (m, 2H, J = 7.2 Hz), 7.06 (s, 1H); 13C NMR (100 MHz CDCl3), δ: 13.94, 22.52, 26.58, 28.11, 29.39, 31.74, 41.01, 50.04, 50.70, 61.23, 169.86.
Preparation of intermediate 6. The protected N-alkyl dipeptide (0.83 g, 2 mmol) was dissolved in 10 mL of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2–trifluoroacetic acid solution. The mixture was stirred at room temperature for 4 h, and then the pH was adjusted to 7 with a 10% Na2CO3 solution. The aqueous phase was extracted with CHCl3 (5 × 10 mL), the organic solution was dried, and the solvent was removed under vacuum. The crude product was purified by chromatography on a silica gel column (CH2Cl2) to give the product as a pale yellow liquid (0.79 g, 99% yield); 1H NMR (400 MHz CDCl3), δ: 0.88 (t, 3H, J = 6.8 Hz), 1.28 (m, 17H, J = 7.8 Hz), 1.72 (s, 2H), 3.06 (s, 2H), 3.16 (m, 1H, J = 3.0 Hz), 3.99 (s, 4H), 3.15 (m, 2H, J = 7.2 Hz), 8.75 (s, 1H); 13C NMR (100 MHz CDCl3), δ: 13.93, 22.58, 25.86, 26.43, 28.97, 29.26, 31.78, 41.18, 45.79, 48.19, 61.43, 80.41, 169.54.
:1 CH2Cl2–trifluoroacetic acid solution. The mixture was stirred at room temperature for 4 h, and then the pH was adjusted to 7 with a 10% Na2CO3 solution. The aqueous phase was extracted with CHCl3 (5 × 10 mL), the organic solution was dried, and the solvent was removed under vacuum. The crude product was purified by chromatography on a silica gel column (CH2Cl2) to give the product as a pale yellow liquid (0.79 g, 99% yield); 1H NMR (400 MHz CDCl3), δ: 0.88 (t, 3H, J = 6.8 Hz), 1.28 (m, 17H, J = 7.8 Hz), 1.72 (s, 2H), 3.06 (s, 2H), 3.16 (m, 1H, J = 3.0 Hz), 3.99 (s, 4H), 3.15 (m, 2H, J = 7.2 Hz), 8.75 (s, 1H); 13C NMR (100 MHz CDCl3), δ: 13.93, 22.58, 25.86, 26.43, 28.97, 29.26, 31.78, 41.18, 45.79, 48.19, 61.43, 80.41, 169.54.
Preparation of target compound (Cu2+ ligand). A 2 M solution of NaOH (10 mL, 20 mmol) was added to a solution of compound 6 (0.8 g, 2 mmol) in MeOH (10 mL). The reaction mixture was stirred for 3 h at room temperature, then the pH of the solution was adjusted to about 7 with 1 M HCl, and the product was collected by filtration as a white solid (0.53 g, 99% yield). 1H NMR (400 MHz D2O), δ: 0.94 (t, 3H, CH3), 1.38 (m, 14H, CH2), 1.74 (m, 2H, CH2), 3.07 (t, 2H, CH2), 3.88 (s, 2H, CH2), 4.03 (s, 2H, CH2); 13C NMR (100 MHz D2O), δ: 13.93, 22.58, 25.86, 26.43, 28.97, 29.26, 31.78, 41.18, 45.79, 48.19, 80.41, 169.54.
Preparation of fluorescent reporter unit in 2 steps
Preparation of fluorescein methyl ester. Concentrated H2SO4 (2.5 mL) was added dropwise to a suspension of fluorescein (3.32 g, 10 mmol) in methanol (10 mL) and the resulting suspension was refluxed for 14 h. The mixture was then cooled, and ice (3 g) followed by NaHCO3 (10 g) was added. The suspension was filtered and washed three times with water and 2% NaHCO3 solution, respectively. The pellet obtained was resuspended in 1% acetic acid, filtered, washed with water, and finally dried at 110 °C for 1 h to give a red powder (3.3 g, 95% yield) without further purification for the next step. 1H NMR (400 MHz CDCl3) δ: 3.94 (s, 3H, CH3), 5.21 (s, 1H, OH), 6.798 (d, 2H, ArH), 6.79 (d, 2H, ArH), 7.30 (d, 2H, ArH), 7.65 (d, 1H, ArH), 7.66 (t, 1H, ArH), 7.71 (t, 1H, ArH), 8.23 (d, 1H, ArH).
Preparation of fluorescent reporter unit. A mixture of ethylene dibromide (197 mg, 91 μL, 1.05 mmol), K2CO3 (288 mg, 2.1 mmol) and N,N-dimethylformamide (DMF, 10 mL) was added to the fluorescein methyl ester (345 mg, 1 mmol), and heated to 60 °C for 3 h. After that, saturated NaCl solution (40 mL) was added to produce a precipitate. The filtered precipitate was washed with water, 1% NaOH solution and water, and finally dried to yield an orange powder without further purification for the next step (443 mg, yield 90%). 1H NMR (400 MHz CDCl3) δ: 3.65 (s, 3H, CH3), 3.68 (t, 2H, CH2), 4.40 (t, 2H, CH2), 6.45 (s, 1H, ArH), 6.54 (d, 2H, ArH), 6.75 (d, 1H, ArH), 6.84 (d, 1H, ArH), 6.90 (d, 1H, ArH), 6.95 (s, 1H, ArH), 7.31 (d, 1H, ArH), 7.67 (t, 1H, ArH), 7.74 (t, 1H, ArH), 8.25 (d, 1H, ArH); 13C NMR (100 MHz CDCl3) δ: 28.25, 52, 44, 68.33, 101.11, 155.88, 113.53, 115.36, 117.91, 129.03, 129.75, 130.05, 130.24, 131.18, 132.75, 134.56, 149.94, 154.1, 158.89, 162.31, 165.58.
Synthesis of Fe3O4 particles. FeCl3·6H2O (0.85 g), trisodium citrate dehydrate (0.48 g), and sodium acetate (1.75 g) were dissolved in ethylene glycol (30 mL) under magnetic stirring. The obtained homogeneous yellow solution was transferred to a Teflon-lined stainless-steel autoclave with a capacity of 40 mL. The autoclave was heated to 200 °C and maintained for 10 h, and then it was cooled to room temperature. The obtained black magnetite particles were washed five times with water and ethanol. The particles were conserved in the ethanol.
Preparation of core–shell Fe3O4@nSiO2 particles. Typically, the Fe3O4 particles (40 mg) were dispersed in an ethanol (200 mL) and water (50 mL) solution, followed by the addition of ammonia aqueous solution (3.0 mL, 25 wt%) and TEOS (200 μL). After stirring at 25 °C for 10 h, the Fe3O4@nSiO2 spheres were obtained and washed three times with water and ethanol, respectively.
Preparation of core–shell Fe3O4@nSiO2@mSiO2 spheres. Typically, the Fe3O4@nSiO2 particles (10 mg) were dispersed in an ethanol (10 mL) and water (30 mL) solution containing 75 mg CTAB, followed by the addition of aqueous ammonia solution (0.27 mL, 25 wt%), TEOS (100 μL) and bis-silylated anthracene (10 μL). After stirring at 25 °C for 10 h, the Fe3O4@nSiO2@mSiO2 spheres were obtained and washed three times with water and ethanol, respectively.
Preparation of Cu2+ fluorescent sensor using Fe3O4@nSiO2 NP as the core. The magnetically recoverable sensor system was prepared by the same procedure with anthracene containing Fe3O4@nSiO2@mSiO2 apart from mixing fluorescein derivative and the ligand in the CTAB ethanol and water solution before the addition of the silica source.
Influence of pH value on the fluorescence emission of the sensor system. The pH value of the fluorescent sensor solution was adjusted using different buffer solutions. The specific operation was as follows: (1) preparation of a series of aqueous solutions with various pH values from 2 to 12 using different buffer solutions; (2) 100 μg of fluorescent Fe3O4@nSiO2@mSiO2 (FFNM) was added to the above solutions (5 mL) and the fluorescent intensity was recorded after equilibrating for 5 min.
Cu2+ detection. The detection procedure was as follows: (1) preparation of 50 μg mL−1 Hepes buffer aqueous solution (50 mM, pH 7.2) of Cu2+ sensor; (2) different amounts of Cu2+ were added to a solution of FFNM-based Cu2+ sensor (3 mL), and the fluorescent intensity was recorded after equilibrating for 5 min.
Adsorption of Cu2+. The experiments to determine the adsorption isotherms were conducted as follows: 2 mg of FFNM was added into a solution which was equilibrated with various concentrations of Cu2+ (10 mL). The concentrations of Cu2+ ranged from 1 to 30 mg L−1 with an initial pH of 7.0.
Disassembly and reassembly of Cu2+ sensor. FFNM-based Cu2+ sensor was dispersed in ethanol for 1 h, then washed with water and magnetically recovered. The disassembly was verified by investigating the I540/I425 ratio. The reassembly was achieved by dispersing FFNM particles in a 5% EtOH water solution containing the ligand and fluorescein, where the addition of a small amount of EtOH was to ensure the dissolution of hydrophobic ligand and fluorescein in water solution.
Characterization
X-ray powder diffraction (XRD) patterns of all samples were recorded on a Rigaku D/MAX-2550 diffractometer using Cu Kα radiation at a wavelength of 0.154 nm, operated at 40 kV and 100 mA. UV-visible absorbance spectra were obtained with a UV-visible spectrophotometer (Varian, Cary 500). The fluorescence spectra of all samples were recorded with a Cary Eclipse fluorescence spectrophotometer. Scanning electron microscopy (SEM) images were obtained with a JEOL JSM-6360LV microscope at an accelerating voltage of 15 kV. Transmission electron microscopy (TEM) images were collected on a JEOL JEM 2010F, and the electron microscope was operated at an acceleration voltage of 200 kV. 1H and 13C NMR spectra were obtained on a Bruker AVANCE DMX500 spectrometer in CDCl3 with tetramethylsilane (TMS) as internal standard. Electron impact mass spectra were recorded on an Agilent 5973N MSD instrument, and ESI mass spectra were recorded on an Agilent 11100-Finnigan instrument. The Zn2+ concentrations were recorded on a Varian-710-ES instrument with ICP/AES data.
Results and discussion
Mesoporous and magnetic characteristics of fluorescent Fe3O4@nSiO2@mSiO2 (FFNM)
Fig. 1 shows TEM images of iron oxide nanoparticles before (A) and after coatings of a nonporous inner silica layer (B, nSiO2) and mesoporous outer silica layer (C and D, mSiO2). The diameter of iron oxide nanoparticles is about 100 nm and the radii of amorphous and mesoporous silica layers are about 35 nm and 50 nm, respectively, where all of particles are highly dispersed. The mesoporous pore system diverges from the center to the fringes (insert of Fig. 1) and has hexagonal mesopore symmetry (Fig. 2C) revealed by HRTEM and small angle XRD pattern, respectively. The wide XRD pattern of FFNM indicates that the iron oxide core is superparamagnetic Fe3O4, and magnetization saturation values of Fe3O4, Fe3O4@nSiO2 and Fe3O4@nSiO2@mSiO2 are 60, 40 and 30 emu g−1, respectively (Fig. 2A, B and D). No remanence was detected in any of the samples according to the hysteresis loop, further confirming the superparamagnetism of these particles.
 |
| | Fig. 1 Fe3O4 nanoparticles (A), Fe3O4@nSiO2 particles (B) and fluorescent Fe3O4@nSiO2@mSiO2 particles (C and D). Insert is an enlargement of the mesoporous shell. | |
 |
| | Fig. 2 Wide XRD patterns of Fe3O4 and Fe3O4@nSiO2 (A), wide XRD pattern of Fe3O4@nSiO2@mSiO2 (B), small angle XRD patterns of Fe3O4@nSiO2 and Fe3O4@nSiO2@mSiO2 (C), and magnetization hysteresis loops of Fe3O4, Fe3O4@nSiO2 and Fe3O4@nSiO2@mSiO2 (D), respectively. | |
Self-assembly of Cu2+ fluorescent sensor
Scheme 1 shows the assembly process of Cu2+ ligand, signal reference and reporter units into the mesoporous shell using CTAB stabilized Fe3O4@nSiO2 NP as the core. We chose bis-silylated anthracene37 and fluorescein derivative as the signal reference and reporter units, which are the energy donor and acceptor of a FRET pair, respectively. They finally occupy the silicate wall, and CTAB occupied the pore channel after the formation of the mesoporous matrix through the electrostatic interaction between cationic surfactant and anionic silicates. Glycylglycine dipeptide is used as the ligand unit,38 and dwells in the hydrophobic micelle with fluorescein derivative attributed to the “like dissolves like” principle. This system functions as a Cu2+ ratiometric sensor through the complex between Cu2+ and its ligand and their influence on the FRET process of the anthracene–fluorescein pair (Scheme 2). The traditional synthesis of a FRET type ratiometric sensor requires a covalent linkage between energy donor and acceptor to achieve effective energy transfer. However, in this system, the covalent linkage between donor and acceptor becomes unnecessary and even the covalent linkage between ligand and fluorophore is avoided, as a result of the effectively shortened distance between the different units confined in the nanometer-sized pore system, since the unit cell parameter of the mesoporous shell is only 4.5 nm.
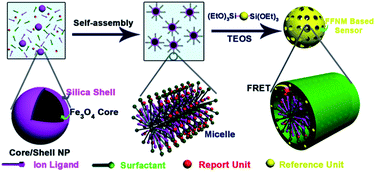 |
| | Scheme 1 Ratiometric fluorescent sensor composed of signal reference unit, signal reporter unit and ligand unit through the formation process of mesoporous silica. | |
 |
| | Scheme 2 Molecular structures of Cu2+ ligand, fluorescent reporter unit and the probable FRET response mechanism to Cu2+. | |
Sensing selectivity to Cu2+
Fig. 3 shows the emission and excitation spectra of anthracene and fluorescein derivatives, where the emission spectrum of anthracene and the absorption spectrum of fluorescein show a good overlap, making it possible for them to form a FRET pair. When the composite assembled from anthracene and fluorescein is excited at 370 nm, where anthracene has obvious absorption while fluorescein has negligible absorption, both emissions from anthracene and fluorescein are observed (Fig. 4A). The above results indicate that an effective FRET process occurs between anthracene and fluorescein even without a covalent linkage.
 |
| | Fig. 3 Emission and excitation spectra of anthracene and fluorescein derivatives. | |
 |
| | Fig. 4 (A) Fluorescence spectra of FFNM-based Cu2+ sensor prepared with different amounts of reporter unit; (B) fluorescence intensity ratios between reporter and reference dyes plotted as I540/I425 at various pH values; (C) responses of sensor to different metal ions (50 μM) and (D) fluorescent response of sensor in the presence of 10 μM Cu2+ and various metal ions (50 μM) (λEx = 370 nm, λEm = 425 and 540 nm). | |
Fig. 4 shows the fluorescence spectra of the FFNM-based Cu2+ sensor prepared with different amounts of reporter unit. The fluorescein concentration is finally fixed at 2 × 10−7 mol for the subsequent study in consideration of the fluorescence self-quenching caused by excessively high molecular concentration (Fig. 4A). Fig. 4B shows the ratiometric (I540/I425) fluorescence response to the variation in pH value, where I540 and I425 refer to the intensity of fluorescein and anthracene, respectively. It is found that the I540/I425 value remains virtually unaffected in the pH range from 6 to 10. The sudden decrease from 5 to 2 should be the result of protonation of fluorescein derivative. Therefore, for subsequent studies, we fixed the pH value at 7.2 using HEPES buffer solution (20 mM). Fig. 4C shows the ratiometric (I540/I425) fluorescence variation of the sensor in the presence of various metal ions. It can be seen that the I540/I425 value dramatically decreases after the addition of Cu2+ (10−5 M). However, the addition of other metal ions such as Fe3+, Pb2+, Cd2+ and Ag+ resulted in negligible change. Fig. 4D shows the interference of other ions such as Li+, Na+, K+, Mg2+, Ca2+, Fe2+, Co2+, Ni2+, Zn2+, Cd2+, Ag+, and Pb2+ (10−4 M) to the I540/I425 value in the presence of Cu2+ (10−5 M). No significant variation of I540/I425 is observed compared with the blank sample with only Cu2+ added, which indicates that this system has an excellent selective detection performance for Cu2+.
Sensitivity of sensor to Cu2+
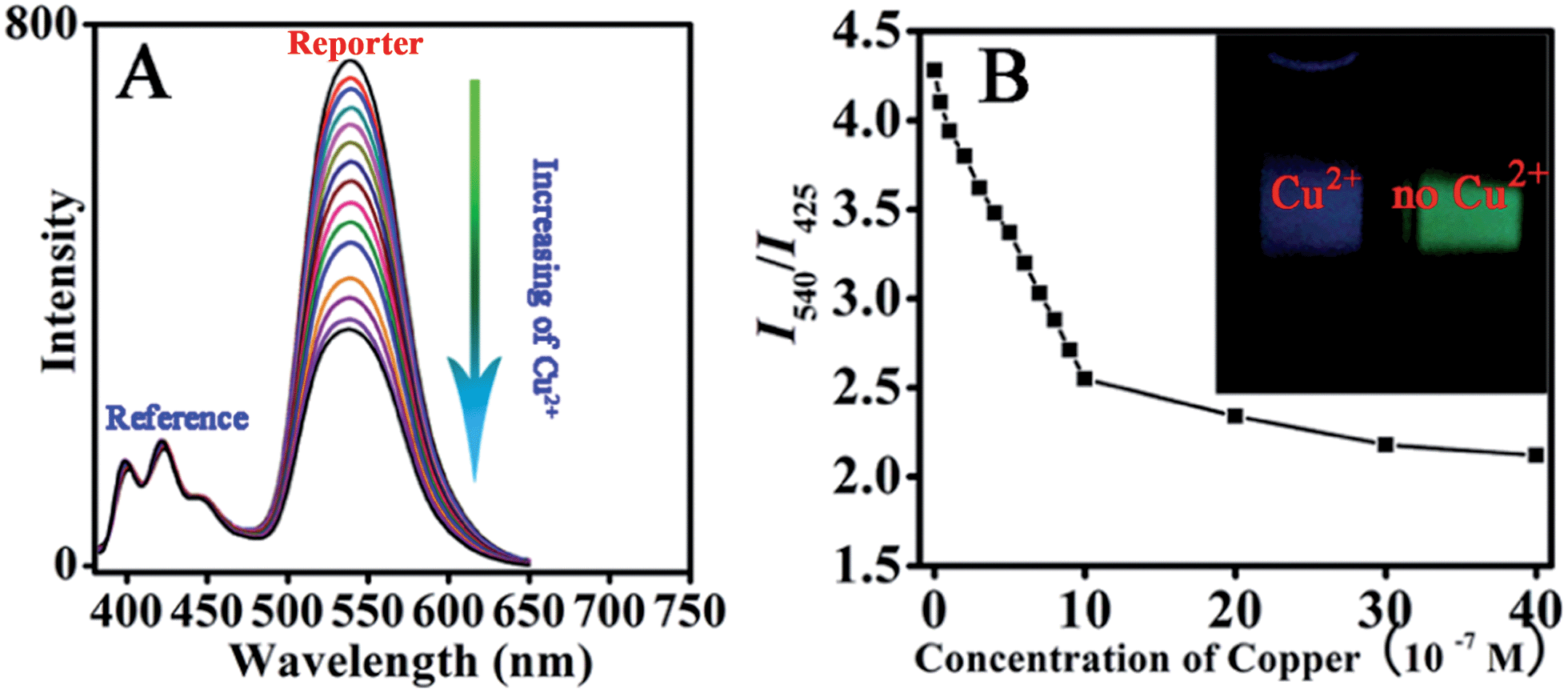
Subsequently, the sensitivity of the above sensor to Cu2+ was investigated, as shown in Fig. 5. It is evident that the emission of the reporter unit at 540 nm gradually decreases with increasing Cu2+ concentration from 0 to 6 × 10−6 M (Fig. 5A). The detection limit is 5 × 10−8 M by setting the confidence level at 3, which indicates that the sensor is highly sensitive. On the other hand, the reference unit shows a stable emission signal, which is hardly affected by the variation in Cu2+ concentration and should be attributed to protection from the framework shell since the encapsulation of anthracene molecules isolates them from the surrounding environment.
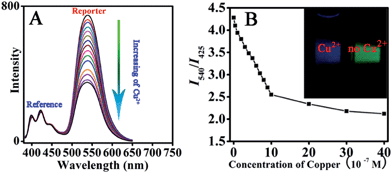 |
| | Fig. 5 Fluorescent spectra of sensor with different amounts of Cu2+ (A); ratiometric calibration curve (I540/I425) as a function of Cu2+ concentration (B) (insert: imaging of sensor solution before and after the addition of Cu2+). | |
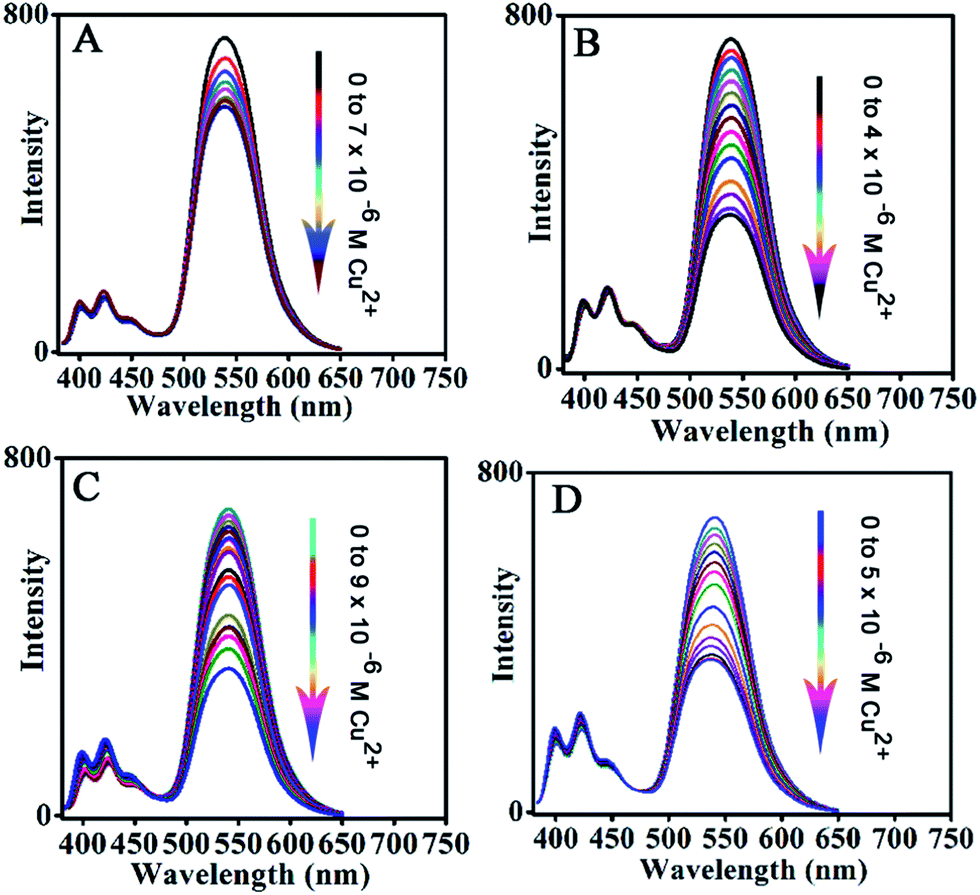
The fluorescence before and after the addition of Cu2+ can be clearly observed with the naked eye under UV light irradiation, as shown in Fig. 5B. The above sensor system with excellent sensing performance is carefully chosen from FFNM particles loaded with different amounts of reporter and ligand units, which are prepared with 2 × 10−7 mol of ligand and 6 × 10−7 mol of signal reporter units (Fig. 6B). Lower and higher amounts of ligand actually lead to decreased sensitivity. The lower sensitivity of the system with a lower amount of receptor (3 × 10−8 mol, Fig. 6A) should be attributed to the ineffective influence of coordinated Cu2+ on remote fluorophores because of the inhomogeneous distribution of the tiny amount of receptor units in the pore channel. On the other hand, the lower sensitivity of the system with a higher amount of ligand (1.2 × 10−6 mol, Fig. 6C) should be attributed to the part isolation of coordinated Cu2+ surrounded by excessive complex units from neighboring fluorophores. Moreover, it is also noted that excessive amounts of reporter unit (Fig. 6D) would also result in decreasing detection sensitivity due to the increasing self-quenching caused by high concentrations of fluorophore.
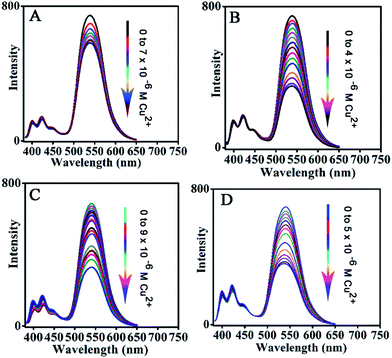 |
| | Fig. 6 Effect of loading amount of fluorescence signal reporter and Cu2+ receptor on detection sensitivity. (A) Ligand (0.1 mL, 3 × 10−4 M), fluorescent reporter (1 mL, 2 × 10−4 M); (B) ligand (2 mL, 3 × 10−4 M), fluorescent reporter (1 mL, 2 × 10−4 M); (C) ligand (4 mL, 3 × 10−4 M), fluorescent reporter (1 mL, 2 × 10−4 M); (D) fluorescent reporter (2 mL, 2 × 10−4 M), ligand (2 mL, 3 × 10−4 M). | |
Adsorption performance
In addition to the sensing performance, we also investigated the adsorption capacity of FFNM sensor for Cu2+, calculated according to the mass balance of Cu2+ ion expressed as eqn (1), where qe is the adsorption capacity (mg g−1), C0 and Ce are the initial and equilibrium concentrations of Cu2+ ions (mg L−1), M is the mass of FFNM (g), and V is the volume of added solution (L).| |
 | (1) |
| |
 | (2) |
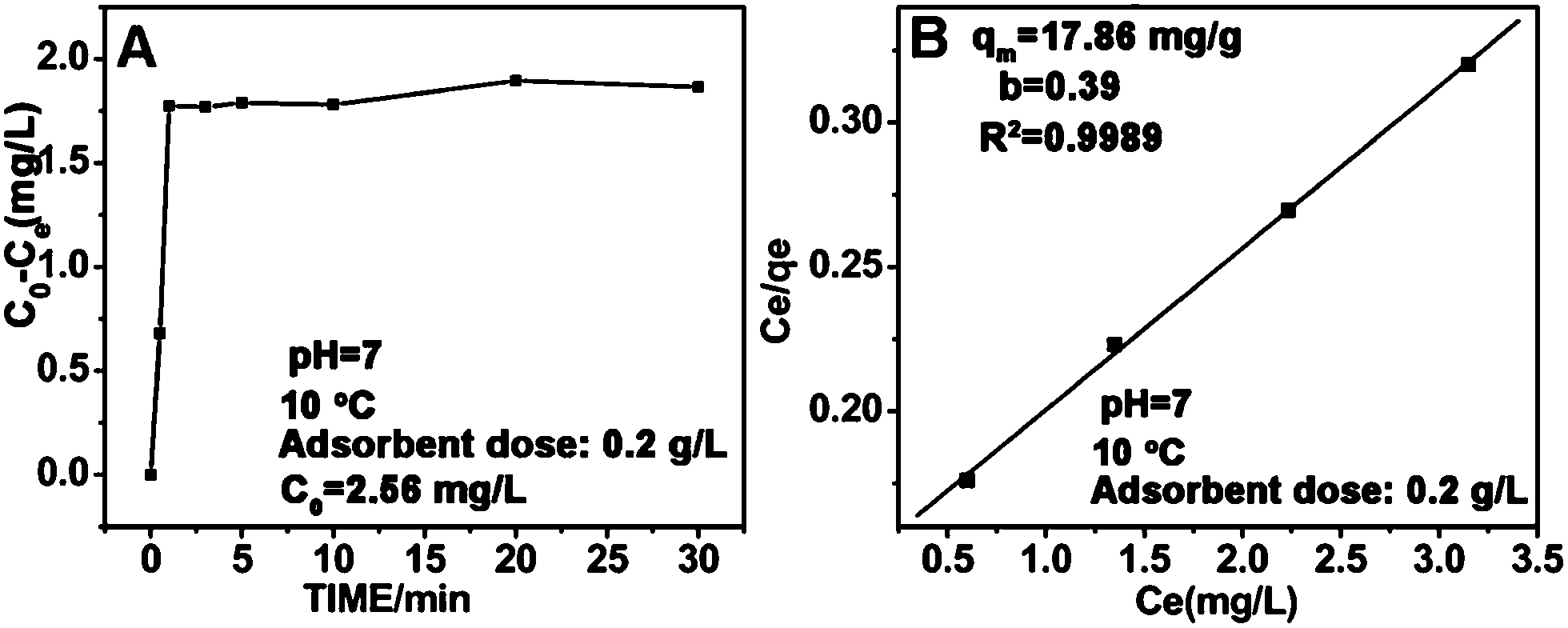
Fig. 7A shows the adsorption kinetics curve of the FFNM-based sensor. It was found that the uptake of Cu2+ by FFNM particles is a very fast process, which finishes in 1 min. This rapid process is attributed to the porous structure of FFNM and the strong affinity of the glycylglycine dipeptide ligand for Cu2+. The adsorption capacity of FFNM for Cu2+ was investigated at different initial metal concentrations (1–30 mg L−1). The adsorption equilibrium data were fitted by the Langmuir adsorption isotherm model as seen from eqn (2), where Ce, qe, b and qm are the equilibrium solution concentration (mg L−1), equilibrium adsorption capacity (mg g−1), Langmuir adsorption equilibrium constant (L mg−1) and maximum adsorption capacity (mg g−1), respectively. The experimental adsorption data were fitted with the Langmuir isotherm equation and are shown in Fig. 7B. It can be seen that the values of b and qm are 0.39 L mg−1 and 17.86 mg g−1, respectively. The R value of 0.9989 means that the adsorption process can be well described by the Langmuir adsorption isotherm, which is dependent on the amount of available adsorption sites on the pore surface of the adsorbent.
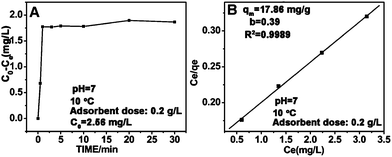 |
| | Fig. 7 Adsorption kinetics curve (A) and Langmuir isotherm curve for the adsorption of Cu2+ by the FFNM sensor (B). | |
Magnetic response and recycling sensing performance
As illustrated above, the FFNM particle is supermagnetic with a saturation magnetization value of 30 emu g−1, and can be rapidly separated from water within 1 min when a magnet is placed near the glass bottle (Fig. 8B). This result indicates that FFNM particles can be easily recovered by applying an external magnetic field after the sensing and adsorption of Cu2+.
 |
| | Fig. 8 Water solution of FFNM particles (A) and their magnetic separation (B). | |
Subsequently, the recycling sensing performance of the FFNM-based sensor was further studied through a disassembly and reassembly process. After the recycling of FFNM by magnetic force, the ligand and signal reporter units in the CTAB micelle were extracted by redispersing FFNM particles in ethanol. The complete extraction was verified by the disappearance of the fluorescence signal of the reporter unit. The I540/I425 value and the sensing ability to Cu2+ can be recovered by dispersing the particles in a 5% ethanol water solution containing signal reporter units and the ligand, where the addition of a small amount of EtOH was to ensure the dissolution of hydrophobic ligand and fluorescein derivative in the water solution. After repeating for 4 cycles, these particles still preserve 90% of the original I540/I425 value and their sensing ability to Cu2+ (Fig. 9). The loss of the fluorescence intensity should be attributed to the loss of CTAB from the pore channel during the ethanol extraction process, leading to the decrease in enrichment ability for ligand and fluorescein.
 |
| | Fig. 9 Reversibility of the fluorescence sensing performance of FFNM to Cu2+ (2 × 10−4 mol) after the disassembly and reassembly of the sensor system. I0 and I′0 refer to the original I540/I425 ratio before and after coordination with Cu2+. I1–I4 correspond to the I540/I425 ratios after the disassembly–reassembly process. I1′–I4′ correspond to the fluorescence intensity of FFNM after reassembly and coordination with Cu2+. | |
Conclusions
We present a novel self-assembly route for the fabrication of a FRET type of ratiometric fluorescent sensor by integrating the preparation process for the sensor into a one-pot formation process of core–shell mesoporous silica using CTAB stabilized Fe3O4@nSiO2 as the core, where all of the units composing the sensor are elaborately arranged in different areas of the mesoporous matrix and can effectively interact with each other even without a direct covalent linkage between them. This nanocomposite system provides a robust platform for the design of novel materials through appropriately utilizing the versatile surfactant chemistry. In this paper, the fabrication and multifunctionalization of the ratiometric fluorescent sensor process is significantly simplified and no effort needs to be paid to covalent bonding of the different sensor units, which should be flexible and variable depending on the detection target.
Acknowledgements
This work has been supported by the National Nature Science Foundation of China (21173077 and 21237003); the National Basic Research Program of China (973 Program, 2013CB632403); the Project of International Cooperation of the Ministry of Science and Technology of China (no. 2011DFA50530); Science and Technology Commission of Shanghai Municipality (12230705000, 12XD1402200); the Research Fund for the Doctoral Program of Higher Education (20120074130001). Open Project from Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control of Nanjing University of Information Science and Technology (kHK1110), Jiangsu Province Innovation Platform for Superiority Subject of Environmental Science and Engineering.
Notes and references
- V. Cauda, A. Schlossbauer, J. Kecht, A. Zurner and T. Bein, J. Am. Chem. Soc., 2009, 131, 11361 CrossRef CAS PubMed.
- G. Büchel, K. Unger, A. Matsumoton and K. Tsutsumi, Adv. Mater., 1998, 10, 1036 CrossRef.
- W. Zhao, J. Gu, L. Zhang, H. Chen and J. Shi, J. Am. Chem. Soc., 2005, 127, 8916 CrossRef CAS PubMed.
- F. Wang, X. L. Chen, Z. X. Zhao, S. H. Tang, X. Q. Huang, C. H. Lin, C. B. Cai and N. F. Zheng, J. Mater. Chem., 2011, 21, 11244 RSC.
- H. Lim and S. Lippard, Acc. Chem. Res., 2007, 40, 41 CrossRef PubMed.
- C. Yang, X. Sun and B. Liu, Anal. Chim. Acta, 2012, 746, 90 CrossRef CAS PubMed.
- Z. Jiang, X. Li, G. Yang, L. Cheng, B. Cai, Y. Yang and J. Dong, Langmuir, 2012, 28, 7174 CrossRef CAS PubMed.
- L. Zhao, S. Zhong, K. Fang and Q. Chen, J. Hazard. Mater., 2012, 239–240, 206 CrossRef CAS PubMed.
- C. Sahin, M. Efecinar and S. Nuray, J. Hazard. Mater., 2010, 176, 672 CrossRef CAS PubMed.
- M. Luconi, R. Olsina, L. Fernandez and M. Silva, J. Hazard. Mater., 2006, 128, 240 CrossRef CAS PubMed.
- A. P. De Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed.
- J. F. Zhang, Y. Zhou, J. Y. Yoon, Y. Kim, S. J. Kim and J. S. Kim, Org. Lett., 2010, 12, 3852 CrossRef CAS PubMed.
- D. Buccella, J. A. Horowitz and S. J. Lippard, J. Am. Chem. Soc., 2011, 133, 4101 CrossRef CAS PubMed.
- K. Hanaoka, Y. Muramatsu, U. Yasuteru, T. Terai and T. Nagano, Chem.–Eur. J., 2010, 16, 568 CrossRef CAS PubMed.
- R. H. Yang, W. H. Chan, A. W. M. Lee, P. F. Xia, H. K. Zhang and K. A. Li, J. Am. Chem. Soc., 2003, 125, 2884 CrossRef CAS PubMed.
- B. C. Zhu, F. Yuan, R. X. Li, Y. M. Li, Q. Wei, Z. M. Ma, B. Du and X. L. Zhang, Chem. Commun., 2011, 47, 7098 RSC.
- X. L. Zhang, Y. Xiao and X. H. Qian, Angew. Chem., Int. Ed., 2008, 47, 8025 CrossRef CAS PubMed.
- S. C. Burdette, C. J. Frederickson, W. W. Bu and S. J. Lippard, J. Am. Chem. Soc., 2003, 125, 1778 CrossRef CAS PubMed.
- Z. P. Liu, W. J. He and Z. J. Guo, Chem. Soc. Rev., 2013, 42, 1568 RSC.
- E. M. Nolan, J. Jaworski, K. Okamoto, Y. Hayashi, M. Sheng and S. J. Lippard, J. Am. Chem. Soc., 2005, 127, 16812 CrossRef CAS PubMed.
- Z. Xu, K. Baek, H. N. Kim, J. N. Cui, X. H. Qian, D. R. Spring, I. Shin and J. Yoon, J. Am. Chem. Soc., 2010, 132, 601 CrossRef CAS PubMed.
- A. Coskun and E. Akkaya, J. Am. Chem.
Soc., 2006, 128, 14474 CrossRef CAS PubMed.
- K. Komatsu, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 13447 CrossRef CAS PubMed.
- A. Mokhir, A. Kiel, D. Herten and R. Kraemer, Inorg. Chem., 2005, 44, 5661 CrossRef CAS PubMed.
- K. M. K. Swamy, S. Ko, S. K. Kwon, H. N. Lee, C. Mao, J. M. Kim, K. Lee, J. Kim, I. Shin and J. Y. Yoon, Chem. Commun., 2008, 5915 RSC.
- L. X. Yan, Z. P. Chen, Z. Y. Zhang, C. G. Qu, L. X. Chen and D. Z. Shen, Analyst, 2013, 138, 4280 RSC.
- P. T. Snee, R. C. Somers, G. J. Nair, P. M. Zimmer, G. D. Bawendi and G. Nocera, J. Am. Chem. Soc., 2006, 128, 13320 CrossRef CAS PubMed.
- M. Frigoli, K. Ouadahi and C. Larpent, Chem.–Eur. J., 2009, 15, 8319 CrossRef CAS PubMed.
- B. Paramanik, S. Bhattacharyya and A. Patra, Chem.–Eur. J., 2013, 19, 5980 CrossRef CAS PubMed.
- B. D. Wang, J. Hai, Z. C. Liu, Q. Wang, Z. Y. Yang and S. H. Sun, Angew. Chem., Int. Ed., 2010, 49, 4576 CrossRef CAS PubMed.
- H. J. Son, H. Y. Lee, J. M. Lim, D. M. Kang, W. S. S. Han, S. J. Lee and H. Jung, Chem.–Eur. J., 2010, 16, 11549 CrossRef CAS PubMed.
- Q. T. Meng, X. L. Zhang, C. He, G. J. He, P. Zhou and C. Y. Duan, Adv. Funct. Mater., 2010, 20, 1903 CrossRef CAS.
- J. L. Liu, C. Y. Li and F. Y. Li, J. Mater. Chem., 2011, 21, 7175 RSC.
- L. X. Mu, W. S. Shi and J. C. Chang, Nano Lett., 2008, 8, 104 CrossRef CAS PubMed.
- X. H. Peng, Y. J. Wang, X. L. Tang and W. S. Liu, Dyes Pigm., 2011, 91, 26 CrossRef CAS PubMed.
- L. Z. Wang, J. Y. Lei and J. L. Zhang, Chem. Commun., 2009, 2195 RSC.
- D. L. Lu, J. Y. Lei, L. Z. Wang and J. L. Zhang, J. Am. Chem. Soc., 2012, 134, 8746 CrossRef CAS PubMed.
- P. Grandini, F. Mancin, P. Tecilla, P. Scrimin and U. Tonellato, Angew. Chem., Int. Ed., 1999, 38, 3061 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.