Remote control of volume phase transition of hydrogels containing graphene oxide by visible light irradiation
Dowan
Kim
a,
Heon Sang
Lee
b and
Jinhwan
Yoon
*a
aDepartment of Chemistry, Dong-A University, Republic of Korea. E-mail: jyoon@dau.ac.kr; Tel: +82 (51) 200-7245
bDepartment of Chemical Engineering, Dong-A University, Republic of Korea
First published on 29th May 2014
Abstract
Poly(N-isopropylacrylamide-co-sodium acrylate) and graphene oxide (GO) composites were prepared by a simple mixing of a solution containing the monomers and aqueous GO through chemically initiated polymerization. We found that exfoliated GO sheets were well-dispersed in the hydrogel matrix and absorbed visible light and consequently generated heat. By combining the photothermal GO with the thermally responsive hydrogel matrix, we demonstrated a volume change of the hydrogel triggered by visible light irradiation of modest intensity. The volume phase transition of the composite hydrogel was fully reversible and could be repeated multiple times without any significant variation. Furthermore, the degree of volume change could be controlled by the light intensity and exposure time as well as the concentration of GO in the hydrogel.
1. Introduction
Hydrogels are three-dimensional elastic networks of hydrophilic polymers formed by physical or chemical cross-linking that can uptake extensive amounts of water in their interstitial space. Because of their elastic structure and high water content, they are useful as membranes,1 food additives,2 drug delivery systems,3,4 and matrices for tissue engineering.5 Hydrogel networks, which consist of stimuli-responsive polymers, can exhibit a reversible volume phase transition in response to external stimuli such as temperature,6,7 pH,8 light,9–19 and an applied magnetic field,20,21 resulting in changes in the polymer–water interactions. Stimuli-responsive behavior is one of the most attractive features of hydrogels and leads to their application in many areas such as sensors,22 actuators,15,18 and microfluidics.11,12,16,17,23 Among these external stimuli for triggering a volume change of hydrogels, light is particularly attractive, because it enables on-demand remote operation with independent local control and rapid switching.To achieve efficient light-responsive hydrogel systems, many studies have focused on immobilizing or entrapping the photoactive materials such as chromophores9–11,24,25 or photothermal materials12–19 with hydrogels. The incorporation of chromophores, which undergo light-induced isomerization or ionization into the hydrogel, has been reported; however, the slow reverse isomerization for spirobenzopyran10,11 or the use of UV light for the azobenzene moiety24,25 may be harmful for biomaterials, somewhat limiting their applicability.
Photothermal conversion materials dispersed in thermally responsive hydrogel matrices have been shown to be efficient for controlling the volume phase transition of hydrogels in response to light.12–19 Crosslinked poly(N-isopropylacrylamide) (PNIPAm), which is a typical temperature responsive hydrogel with a lower critical solution temperature of around 32 °C, has been frequently used as a thermally responsive matrix. When heated in water, PNIPAm-based hydrogels undergo a reversible volume phase transition from a swollen hydrated state to a shrunken dehydrated state.26 Photothermal materials in the hydrogel matrix absorb light and consequently generate heat, thus triggering a volume transition of the thermally responsive hydrogel matrix.
It is possible to further categorize photothermal materials according to the wavelength of light by which they are activated. Most studies on these systems dealt with near-infrared (NIR)-sensitive materials such as gold nanoparticles,12,13 single wall carbon nanotubes,14,15 and graphene oxide (GO).16–18 These materials are potentially better suited to biomedical applications, as most biomaterials are transparent to and not damaged by NIR radiation. Although effective for biomedical applications, a high-intensity laser source (i.e., several W cm−2) is required to cause the volume change of the hydrogels by a photothermal effect.12–18 Furthermore, for some potential applications using light-responsive hydrogels, such as smart windows for sunlight protection and drug release systems for skin therapy, visible light of a modest intensity would be a more appropriate trigger. Despite these requirements, only a few investigations have been devoted to visible light-responsive hydrogels using a photothermal effect.19
Motivated by the advantages of GO, that is, abundance, low cost and biocompatible,27 in this study, we developed thermally responsive PNIPAm-based hydrogel and aqueous GO composites that undergo reversible changes in the swelling ratio in response to visible light exposure. We note that several researchers have reported volume changes of hydrogel and GO composites triggered by NIR light.16–18 However, according to a previous report, GO may be decomposed or deoxygenated by high-intensity NIR light, depending on the power and exposure time.28 This may lead to changes in the photothermal efficiency of GO, thereby making reliable and precise volume change of hydrogels difficult. Here, we instead used a modest intensity of visible light to trigger the fully reversible volume phase transition of the hydrogel by the photothermal effect of GO.
Moreover, we prepared the GO/hydrogel composites by simple mixing of the solution containing the monomers with aqueous GO through a chemically initiated polymerization, whereas other studies used covalent bonding between GO and the polymer16 or γ-irradiation-assisted polymerization.17 Through this simple and straightforward method, we realized a visible light-responsive hydrogel that exhibited a fully reversible and repeatable volume phase transition.
2. Experimental
2.1 Materials
Aqueous GO (5 mg mL−1, composition: carbon (79%); oxygen (20%), flake size: 0.5–5 μm) was obtained from Graphene Supermarket (Calverton, NY, USA). N-isopropylacrylamide (NIPAm) was purchased from TCI (Nihonbashi-honcho, Chuo-ku, Japan). Phosphate buffered saline (PBS) was obtained from Bio Basic Inc. (Markham Ontario, Canada). Aqueous fluorescent polystyrene beads of 3 μm were obtained from PolyScience Inc. (Warrington, PA, USA). All other chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA) and used as received without further purification.2.2 Preparation of the GO/hydrogel composites
The composite solutions were prepared by a simple mixing of aqueous GO with the monomer solutions, thus entrapping the GO sheets within the hydrogel matrix after polymerization. A monomer solution (50 μL) containing 7.21 mg (637.2 mM) NIPAm, 0.19 mg (19.9 mM) sodium acrylate (NaAc), and 0.10 mg (6.5 mM) N,N′-methylenebis(acrylamide) (BisAA) was mixed with 50 μL of a 5 mg mL−1 aqueous dispersion of GO, which corresponds 3.3 wt% to the monomer content. A dilute suspension (0.3 μL) of fluorescent polystyrene beads in water was then added to the mixed solution. The concentration of the fluorescent beads was determined by yield that average of ∼10 to 20 beads per ∼1 mm2. Free radical polymerization was initiated by adding 0.3 μL of N,N,N′,N′-tetramethylethylenediamine and 0.6 μL of a 10 wt% ammonium persulfate aqueous solution to 100 μL of degassed composite solution. The resulting solutions were immediately loaded into a capillary channel formed with two coverslips separated by spacers of 140 μm. Gelation was carried out in a sealed chamber under a positive pressure of nitrogen for 1 h. After polymerization, the coverslip and spacers were removed, and the hydrogel was immersed in PBS (137 mM NaCl) swelling medium. To extract unreacted components, the swelling medium was changed at least three times over a period of 3 h.2.3 Measurements
To measure the swelling ratios of the composite hydrogels, the positions of a collection of at least 10 embedded fluorescent beads within the hydrogel were tracked as a function of irradiation time, as described previously.29 The change of the linear swelling ratio is identical to the movement of the fluorescent beads. The reported values are the average of the linear expansion determined for each bead, with the uncertainties being the standard deviation.Blue light irradiation was provided by exposing samples to light from a high-pressure mercury short arc lamp (EL-6000, Leica) through a blue excitation filter (450–490 nm, I3, Leica), which yielded an intensity of 45 mW cm−2, as measured by an ACCU-CAL™ 50-LED (DYMAX). The swelled hydrogel films were imaged using an epi-fluorescence microscope (DMI3000B, Leica). X-ray diffraction measurements were performed using an Ultima IV (Rigaku) with Cu Kα radiation.
3. Results and discussion
Fig. 1 shows the temperature changes of aqueous GO and water under visible light irradiation. We found that GO sheets dispersed in water absorbed visible light of 45 mW cm−2 and consequently generated heat. As seen in Fig. 1, the temperature of the aqueous GO increased from 23.2 °C to 29.4 °C when irradiated with visible light for 3 min. In contrast, the pure water did not undergo a significant temperature change upon irradiation with visible light. These results indicate that sufficient heat is produced under visible light irradiation to cause a substantial temperature increase of aqueous GO, meaning that the aqueous GO dispersion converts the photo energy of visible light into thermal energy, which increases the temperature of the water.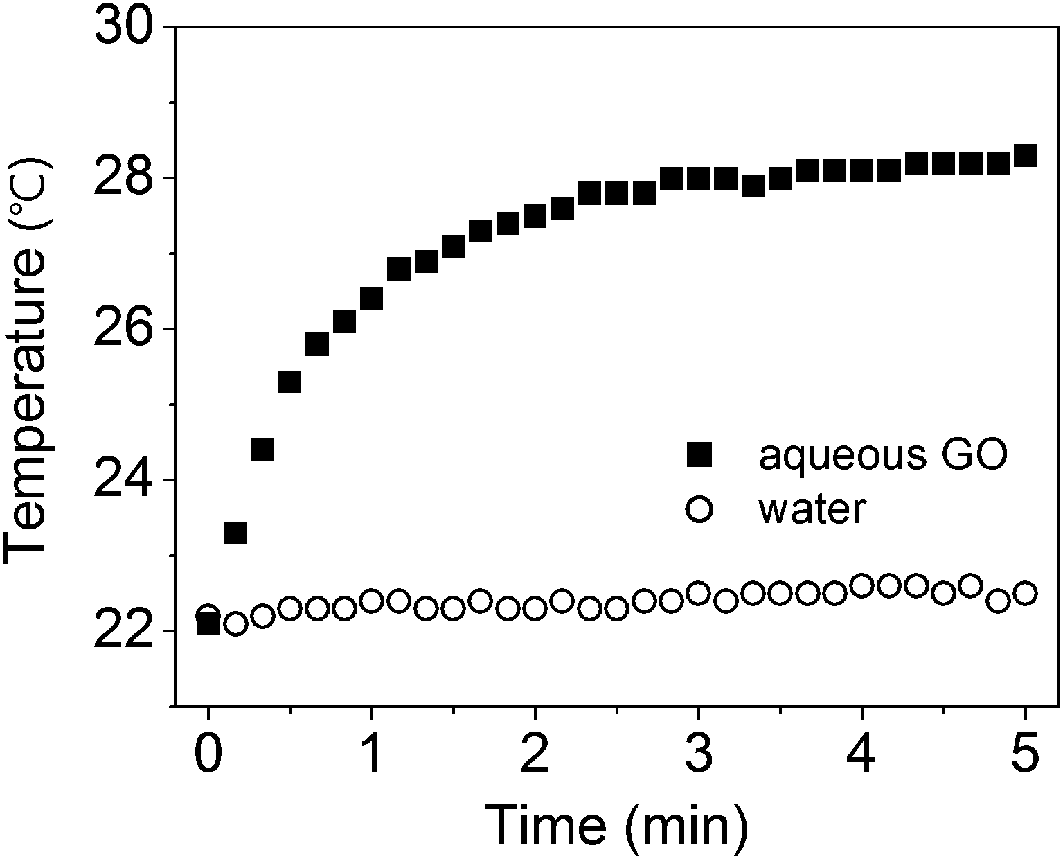 | ||
| Fig. 1 Temperature changes for pure water and 2.5 mg mL−1 aqueous graphene oxide (GO) under irradiation with visible light. | ||
To develop visible light-responsive hydrogels, we attempted to prepare temperature-responsive poly(N-isopropylacrylamide-co-sodium acrylate) copolymer (p(NIPAm-co-NaAc)) and aqueous GO composites through chemically initiated free-radical polymerization. As mentioned above, the volume phase transition can be expected in response to visible light by combining the thermally responsive hydrogel network with photothermal materials. GO can be stably dispersed in water in its exfoliated form of monolayer sheets due to the hydrophilic oxygen-containing surface functional groups.30 The composite solutions were formed by mixing the aqueous GO with the NIPAm-monomer solution containing 3 mol% NaAc, thus entrapping the GO sheets within the hydrogel matrix after polymerization. The pure p(NIPAm-co-NaAc) was colorless and transparent, while the p(NIPAm-co-NaAc)/GO composite hydrogel was pale brown-black but still transparent.
Fig. 2 shows the XRD profiles for pure GO, p(NIPAm-co-NaAc), and p(NIPAm-co-NaAc) containing GO. The characteristic diffraction peak of GO was observed at 2θ = 10.8°, corresponding to the interlayer spacing of stacked GO sheets, while two broad peaks were observed at 2θ = 7.9° and 21.5° for freeze-dried p(NIPAm-co-NaAc). After GO was dispersed into the p(NIPAm-co-NaAc) matrix through polymerization, the XRD profile for the composite showed only two broad peaks corresponding to the p(NIPAm-co-NaAc) diffraction pattern. The typical GO peak completely disappeared, suggesting that the exfoliated GO sheets were well-dispersed in the p(NIPAm-co-NaAc) matrix.17 By simple mixing of each aqueous solution and chemical initiation of the radical polymerization, which does not require any radiation source for initiation, we could prepare the hydrogel composite containing well-dispersed GO.
 | ||
| Fig. 2 XRD profiles for dried GO, poly(N-isopropylacrylamide-co-sodium acrylate) (p(NIPAm-co-NaAc)), and p(NIPAm-co-NaAc) containing 3.3 wt% GO. | ||
To compare the thermal behavior of p(NIPAm-co-NaAc) gel containing GO with a pure hydrogel of the same chemical composition, the linear swelling ratios are plotted as a function of temperature in Fig. 3. The equilibrium linear swelling ratio, λf, is defined as the amount a gel swells in each dimension at the equilibrium state when immersed in water, meaning that the volume–swelling ratio is equal to λf.3
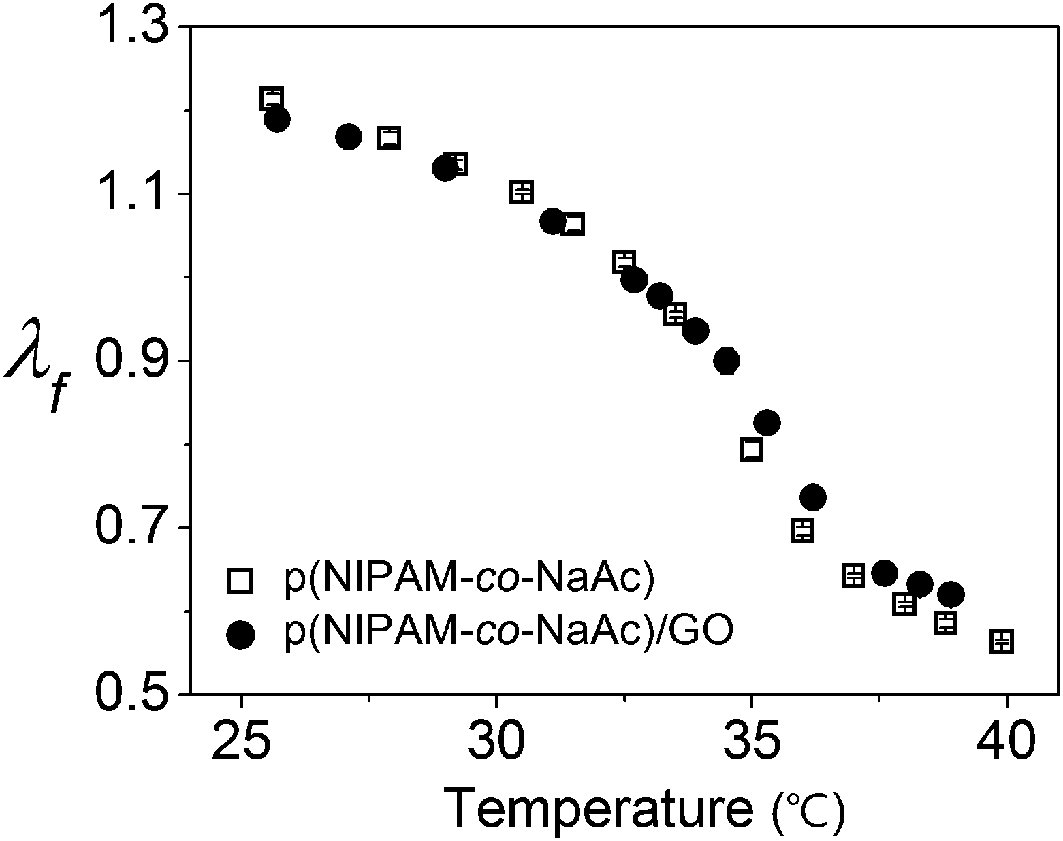 | ||
| Fig. 3 Temperature-dependent linear swelling ratio λf during heating for pure p(NIPAm-co-NaAc) and p(NIPAm-co-NaAc) containing 3.3 wt% GO. To measure the equilibrium swelling ratio, the temperature was held for 15 min prior to determining the degree of swelling. | ||
As shown in Fig. 3, the hydrogel containing GO shows nearly the same thermal behavior as that of the pure hydrogel of the same chemical composition. This result reveals that the GO contents used in this work have little influence on the thermo-responsivity of the composite hydrogel.
As shown in previous reports,6,7 p(NIPAm-co-NaAc) gel undergoes a reversible volume phase transition from a swollen hydrated state to a shrunken dehydrated state with an increase in temperature. To yield a linear variation in the swelling over the temperature range of the photothermal effect, from 20 °C to 30 °C, 3 mol% NaAc was copolymerized with NIPAm. It is essential to have a linear variation in the swelling of hydrogel matrix as a function of temperature rather than a discontinuous volume phase transition to guarantee reliable and controllable volume changes by heat generated from the photothermal effect.
By combining the thermally responsive p(NIPAm-co-NaAc) matrix of the composite hydrogel and the photothermal GO dispersed in the matrix, we demonstrated a reversible volume change of the composite hydrogel under visible light exposure. Fig. 4 shows the equilibrium swelling ratio changes of p(NIPAm-co-NaAc) containing GO under visible light irradiation. As seen in Fig. 4, p(NIPAm-co-NaAc) containing GO at the equilibrium swelling state shows a linear swelling ratio (λf) of 1.22. When exposed to blue light of 45 mW cm−2, the hydrogel/GO composites underwent significant volume changes proportional to the exposure time. For the sample under 1 min irradiation, the linear swelling ratio decreases to 1.15, while the linear swelling ratio of the same sample is 1.07 after 3 min irradiation. Irradiation with visible light for 5 min led to the decrease of the linear swelling ratio to 1.01, which corresponds to an approximately 43% decrease in the volume compared to the initial equilibrium state before light exposure. Based on the measured linear swelling ratio, we further investigated the degree of volume change depending on the irradiation time and plotted the results in the inset of Fig. 4. We found that the decrease in volume is linearly proportional to the irradiation time, suggesting that the degree of volume change can be easily manipulated by controlling the light exposure time.
 | ||
| Fig. 4 Linear swelling ratios λf for p(NIPAm-co-NaAc) containing 3.3 wt% GO exposed to 45 mW cm−2 of blue light for 1, 3, and 5 min. Inset: visible light-induced volume shrinkage percent, which was calculated from the linear swelling ratios λf. | ||
Soon after turning the visible light off, the hydrogel/GO composite started to recover its original volume by reswelling. The swelling ratio increased gradually back to its former state, and the initial equilibrium volumes were recovered in 6–7 min after the light was turned off, regardless of the degree of volume shrinkage. These results are consistent with the theory for hydrogel swelling kinetics, which depends only on their dimensions.29
Two control experiments were also repeated for the same amount of aqueous GO in non-responsive poly(acrylamide) (PAAm) hydrogel and for pure p(NIPAm-co-NaAc) hydrogel without GO. The volumes of both hydrogels were maintained even after 10 min of visible light exposure (data not shown).
Furthermore, we found that this reversible volume change of the composite hydrogel triggered by visible light could be repeated multiple times without significant variation. The linear swelling ratios were measured for 3 min irradiation followed by a recovery time of 15 min, and this cycle was repeated 20 times. As seen in Fig. 5, the composite hydrogel exhibited a fully reversible volume change at each cycle with constant and reliable volume changes. This result shows that no aging or degradation of the GO in the hydrogel matrix occurs, which may cause a change in the photothermal efficiency. According to a previous report, the photothermal effect of GO can be altered by being reduced or degraded by irradiation with high-intensity light.
 | ||
| Fig. 5 Visible light-induced change in the swelling ratio, which was found to be reversible and repeatable over 20 cycles. Each cycle consists of light illumination for 3 min followed by a recovery time of 15 min. | ||
We also found that degree of volume change for the composite hydrogels can be customized by changing the concentration of GO and light intensity, as well as the irradiation time. As shown in Fig. 6, a greater loading of GO or stronger light intensity induces a greater volume change under otherwise identical conditions, suggesting that the concentration of GO in the hydrogel and the intensity of light can be used to effectively control the degree of volume change.
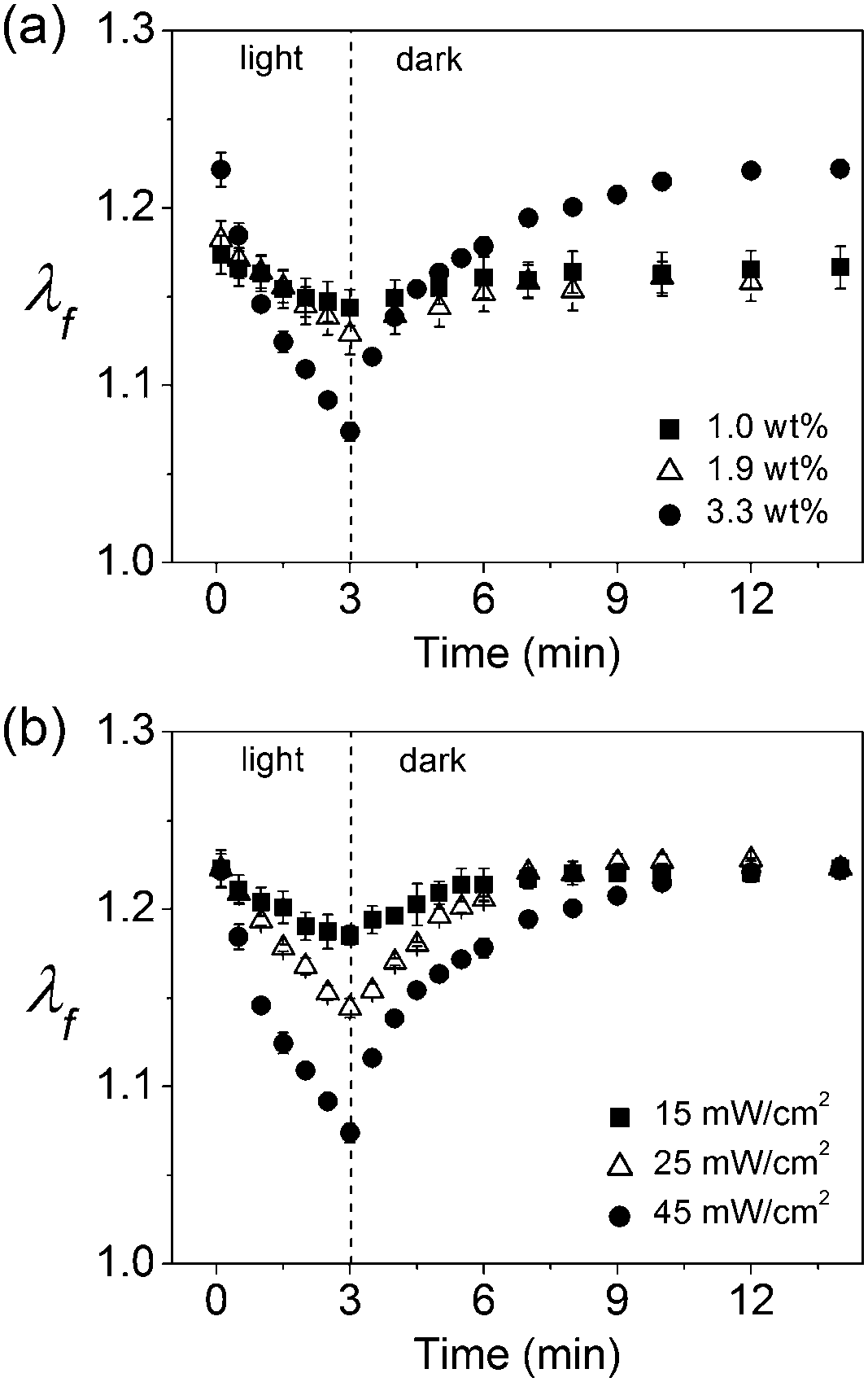 | ||
| Fig. 6 (a) Linear swelling ratios λf for p(NIPAm-co-NaAc) containing 1.0, 1.9, and 3.3 wt% GO exposed to 45 mW cm−2 of blue light for 3 min. (b) Linear swelling ratios λf for p(NIPAm-co-NaAc) containing 3.3 wt% GO exposed to 15, 25, and 45 mW cm−2 of blue light for 3 min. | ||
To confirm that photothermal conversion by GO is responsible for the light-induced volume phase transition of the hydrogels, we attempted to take thermal images of the composite hydrogel under irradiation conditions. As seen in Fig. 7, the surface temperature of the hydrogel was constant over the whole region at the equilibrium swelling state. When a specific region was irradiated with visible light, the thermal image of the hydrogel surface showed a significant increase in the temperature of the irradiated region. As estimated from the thermal image in Fig. 7, the surface temperature of only the irradiated region rises from an initial temperature of ∼25 °C to ∼29 °C after a 3 min irradiation. In contrast, a negligible temperature increase was observed in the area surrounding the irradiated region. After turning off the light, the surface temperature decreased to its initial value, confirming that the light-induced volume change of the composite hydrogel arises from the coupling of the photothermal conversion by GO to the temperature-responsive PNIPAm copolymer gel. We note that the hydrogel was exposed to the air for imaging contrast enhancement during the acquisition of the thermal images; thus, the temperature rise could be underestimated owing to the heat transfer to the air by convection.
 | ||
| Fig. 7 Thermal photographs of p(NIPAm-co-NaAc) containing 3.3 wt% GO before (left) and after (right) exposure to 45 mW cm−2 of blue light for 3 min. The white dotted circle in the right photograph denotes the beam size. | ||
4. Conclusion
We can conclude that composite hydrogels containing photothermal conversion materials embedded within a thermally responsive polymer matrix provide an efficient means to trigger changes in volumetric swelling by visible light irradiation of modest intensity. We found that the volume change of a hydrogel composite was induced by the temperature response of p(NIPAm-co-NaAc) to thermal energy converted from the photo energy of visible light by GO. The incorporated GO in the p(NIPAm-co-NaAc) matrix absorbed the visible light and converted the photo energy into thermal energy, which heated up the hydrogel network. The degree of volume change was fully reversible and repeatable over multiple cycles, indicating that the photothermal conversion efficiency of GO was maintained. This responsiveness could be triggered remotely and provided flexible control by varying the light intensity and exposure time.Acknowledgements
This research was supported by the Ministry of Education (MOE), Ministry of Science, ICT and Future Planning (MSIP), and the National Research Foundation of Korea (NRF) through the Human Resource Training Project for Regional Innovation (2012H1B8A2025809) and Basic Research fund (2011-0015029).References
- R. T. Swank and K. D. Munkres, Anal. Biochem., 1971, 39, 462–477 CrossRef CAS.
- X. Chen, B. D. Martin, T. K. Neubauer, R. J. Linhardt, J. S. Dordick and D. G. Rethwisch, Carbohydr. Polym., 1995, 28, 15–21 CrossRef CAS.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS PubMed.
- N. A. Peppas, K. B. Keys, M. Torres-Lugo and A. M. Lowman, J. Controlled Release, 1999, 62, 81–87 CrossRef CAS.
- X. Z. Shu, Y. Liu, F. S. Palumbo, Y. Luo and G. D. Prestwich, Biomaterials, 2004, 25, 1339–1348 CrossRef CAS PubMed.
- S. Hirotsu, Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1987, 87, 1392–1395 CrossRef CAS PubMed.
- J. Yoon, J. Kim and R. C. Hayward, Soft Matter, 2010, 6, 5807–5816 RSC.
- O. E. Philippova, D. Hourdet, R. Audebert and A. R. Khokhlov, Macromolecules, 1997, 30, 8278–8285 CrossRef CAS.
- A. Suzuki and T. Tanaka, Nature, 1990, 346, 345–347 CrossRef CAS.
- K. Sumaru, K. Ohi, T. Takagi, T. Kanamori and T. Shinbo, Langmuir, 2006, 22, 4353–4356 CrossRef CAS PubMed.
- S. Sugiura, K. Sumaru, K. Ohi, K. Hiroki, T. Takagi and T. Kanamori, Sens. Actuators, A, 2007, 140, 176–184 CrossRef CAS PubMed.
- S. R. Sershen, G. A. Mensing, M. Ng, N. J. Halas, D. J. Beebe and J. L. West, Adv. Mater., 2005, 17, 1366–1368 CrossRef CAS.
- T. Kawano, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Bioconjugate Chem., 2009, 20, 209–212 CrossRef CAS PubMed.
- T. Fujigaya, T. Morimoto, Y. Niidome and N. Nakashima, Adv. Mater., 2008, 20, 3610–3614 CrossRef CAS.
- X. Zhang, C. L. Pint, M. H. Lee, B. E. Schubert, A. Jamshidi, K. Takei, H. Ko, A. Gillies, R. Bardhan, J. J. Urban, M. Wu, R. Fearing and A. Javey, Nano Lett., 2011, 11, 3239–3244 CrossRef CAS PubMed.
- C.-W. Lo, D. Zhu and H. Jiang, Soft Matter, 2011, 7, 5604–5609 RSC.
- C.-H. Zhu, Y. Lu, J. Peng, J.-F. Chen and S.-H. Yu, Adv. Funct. Mater., 2012, 22, 4017–4022 CrossRef CAS.
- E. Wang, M. S. Desai and S.-W. Lee, Nano Lett., 2013, 13, 2826–2830 CrossRef CAS PubMed.
- J. Yoon, P. Bian, J. Kim, T. J. McCarthy and R. C. Hayward, Angew. Chem., Int. Ed., 2012, 51, 7146–7149 CrossRef CAS PubMed.
- N. S. Satarkar and J. Z. Hilt, Acta Biomater., 2008, 4, 11–16 CrossRef CAS PubMed.
- N. S. Satarkar and J. Z. Hilt, J. Controlled Release, 2008, 130, 246–251 CrossRef CAS PubMed.
- Z. Shakhsher, W. R. Seitz and K. D. Legg, Anal. Chem., 1994, 66, 1731–1735 CrossRef CAS.
- N. S. Satarkar, W. Zhang, R. E. Eitel and J. Z. Hilt, Lap Chip, 2009, 9, 1773–1779 RSC.
- Y.-L. Zhao and J. F. Stoddart, Langmuir, 2009, 25, 8442–8446 CrossRef CAS.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef CAS PubMed.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl. Chem., 1968, 2, 1441–1455 CrossRef CAS.
- K. Wang, J. Ruan, H. Song, J. Zhang, Y. Wo, S. Guo and D. Cui, Nanoscale Res. Lett., 2011, 6(8), 1–8 Search PubMed.
- V. Abdelsayed, S. Moussa, H. M. Hassan, H. S. Aluri, M. M. Collinson and M. S. El-Shall, J. Phys. Chem. Lett., 2010, 1, 2804–2809 CrossRef CAS.
- J. Yoon, S. Cai, Z. Suo and R. C. Hayward, Soft Matter, 2010, 6, 6004–6012 RSC.
- W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. Yang, A. Velamakanni, S. J. An, M. Stoller, J. An, D. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |