Facile synthesis of dimeric BODIPY and its catalytic activity for sulfide oxidation under visible light†
Liang Wangab,
Jing Caoab,
Jian-wei Wangab,
Qun Chenab,
Ai-jun Cui*ab and
Ming-yang He*ab
aJiangsu Key Laboratory of Advanced Catalytic Materials and Technology, Changzhou University, 213164, P. R. China
bSchool of Petrochemical Engineering, Changzhou University, Changzhou 213164, P. R. China. E-mail: lwcczu@126.com; hemingyangjpu@yahoo.com; Fax: +86-519-86330251; Tel: +86-(0)13961477422
First published on 17th March 2014
Abstract
An orthogonal dimeric BODIPY was easily prepared via condensation of 2,4-dimethylpyrrole and oxalyl dichloride, and was utilized as a visible-light-driven photocatalyst for the oxidation of sulfides. High catalytic efficiencies, and mild and green conditions are the major advantages of this protocol. Moreover, meso-carbalkoxylated BODIPYs could also be prepared using a similar one-pot condensation of 2,4-dimethylpyrrole, oxalyl dichloride and a series of alcohols.
Photoredox catalytic organic reactions driven by visible light have been gaining increasing interest due to the mild conditions for substrate activation, leading to the construction of complex organic compounds with a feasible synthetic method.1 However, the potential toxicity, high cost as well as the limited availability of the current organometallic photocatalysts are the major drawbacks. Thus, looking for a metal-free, readily available or easily prepared, and green photocatalysts is still a challenge in this field.
Boron–dipyrromethene (BODIPY) compounds have received much attention because of their unique properties such as high fluorescence quantum yields (Φf), large molar absorption coefficients (ε), excellent thermal and photochemical stabilities.2–4 Much effort in decoration of the BODIPY scaffold with reactive functionalities either at meso-position or 8-position have been realized for tuning their fluorescence characteristics,5–11 which provides them a prominent place as outstanding fluorophores for use in fluorescent materials, labels and probes.12–14 Besides, BODIPY derivatives have shown good photocatalytic activities including oxidations, cross-dehydrogenative coupling reactions as reported by Ramaiah group, Jing group as well as Zhao group.15–19 Comparing with the conventional photocatalysts such as Ru(bpy)3Cl2 (bpy = 2,2′-bipyridine) or Nile Red, BODIPY derivatives are less-toxic and low cost, but with strong absorption of visible light, long-lived triplet excited state and readily tunable molecular structures. To our knowledge, the reported BODIPY photocatalysts are focused on iodo-BODIPYs since fluorophores bearing heavy atom generally have a high intersystem crossing quantum yield (Φisc) and a high singlet oxygen quantum yield (Φ△) due to the heavy atom effect.20,21 Recently, Akkaya et al. designed two kinds of orthogonal dimeric BODIPYs with respectable singlet oxygen quantum yields and increased intersystem crossing but without heavy atoms.11 Their applications as photocatalysts in organic reactions, however, were not reported.
Herein, we wish to report a modified and facile route for the synthesis of orthogonal dimeric BODIPY (1, Fig. 1) and its application for the oxidation of sulfides.
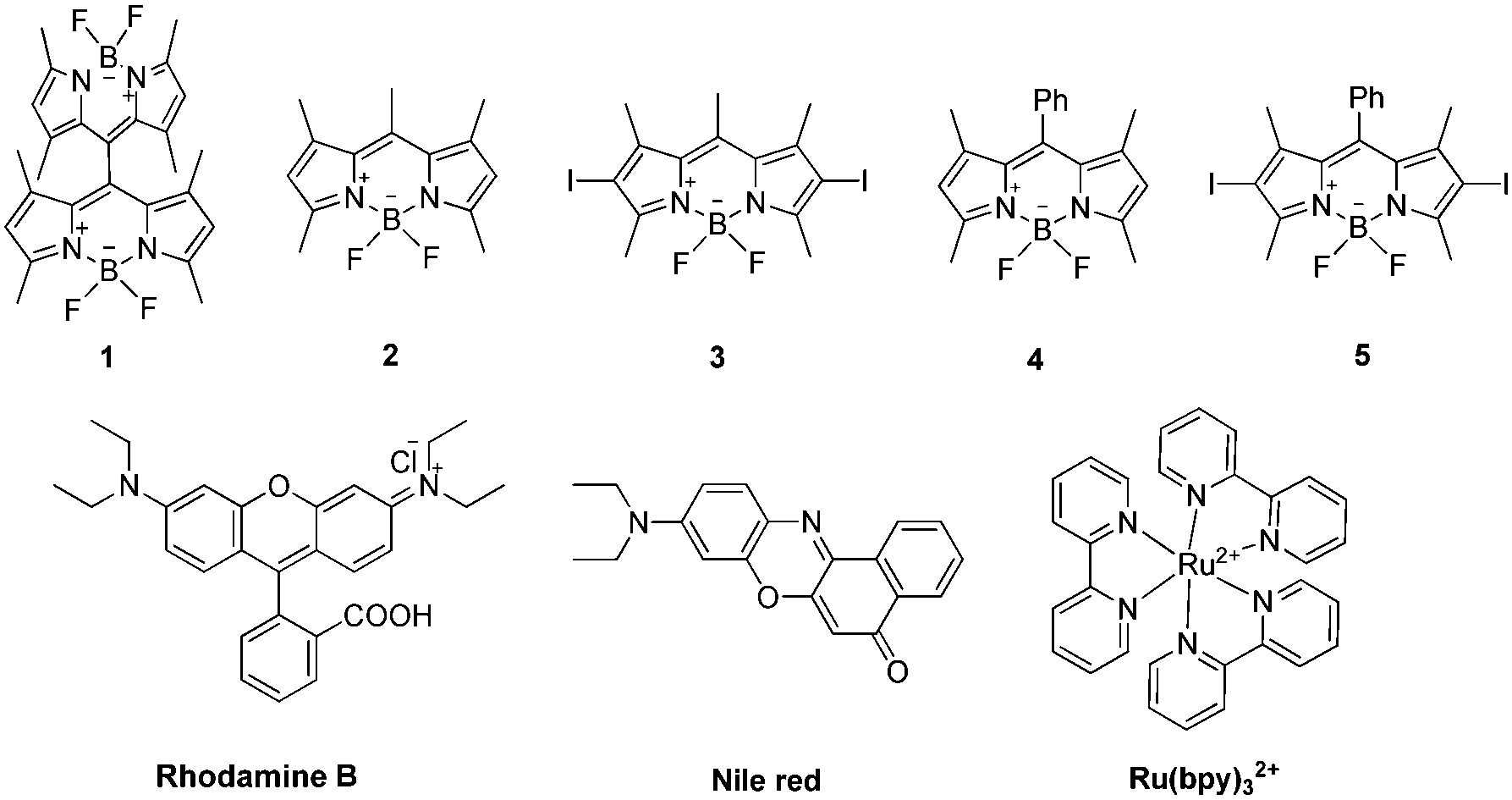 | ||
| Fig. 1 The orthogonal dimeric BODIPY and other photocatalysts surveyed in this study. | ||
An initial investigation focused on the preparation of dimeric BODIPY 1 (Scheme 1). The reported methods generally involved the following steps: (1) condensation of acetoxyacetyl chloride and 2 equiv. of 2,4-dimethyl pyrrole under reflux in dichloromethane followed by treatment of the reaction mixture with 4 equiv. of BF3·OEt2 and diisopropylethylamine; (2) hydrolysis under basic conditions; (3) oxidation to the corresponding meso-formyl BODIPY using standard Dess–Martin oxidation conditions; and (4) standard BODIPY synthetic progress. It was obvious that this route was long and several purification processes were required, while a 7.2% total yield was obtained.11,22 In our study, 2,4-dimethylpyrrole and oxalyl dichloride were selected as starting materials and it was pleased to obtain the dimeric BODIPY 1 in 10% yield via only one-step condensation.
 | ||
| Scheme 1 Preparation of dimeric BODIPY 1. | ||
Further studies showed that a series of carbalkoxylated BODIPYs could be prepared via the one-pot condensation of 2,4-dimethylpyrrole, oxalyl dichloride and substituted alcohols, affording the BODIPYs 6–10 in satisfactory yields. This is due to the fact that acyl chloride is more active than the aldehyde and no oxidation process is required during the first step. To the best of our knowledge, this is the first report for the preparation of carbalkoxylated BODIPYs. Replacing the alcohols with amines, however, provided no products (Scheme 2).
 | ||
| Scheme 2 Preparation of carbalkoxylated BODIPY 6–10. | ||
The photophysical properties of BODIPY 1 and 6–10 were tested. For BODIPY 1, the maximum wavelengths of absorption and emission in CH2Cl2 were 515 and 606 nm, respectively (Fig. 2), which was much higher than carbalkoxylated BODIPYs 6–10. This was attributed to the high conjugation of dimeric BODIPY 1. The photophysical properties of BODIPY 1 in other solvents gave obvious differences and the largest stocks shift (∼102 nm) was found in methanol, while its fluorescence quantum yield was much lower (0.004) than that in hexane (0.721). For BODIPYs 6–10, generally, relatively lower fluorescence quantum yields were obtained. This was due to the strong electron-withdrawing effect of alkoxycarbonyl groups at the meso-position, leading to decreased fluorescence.
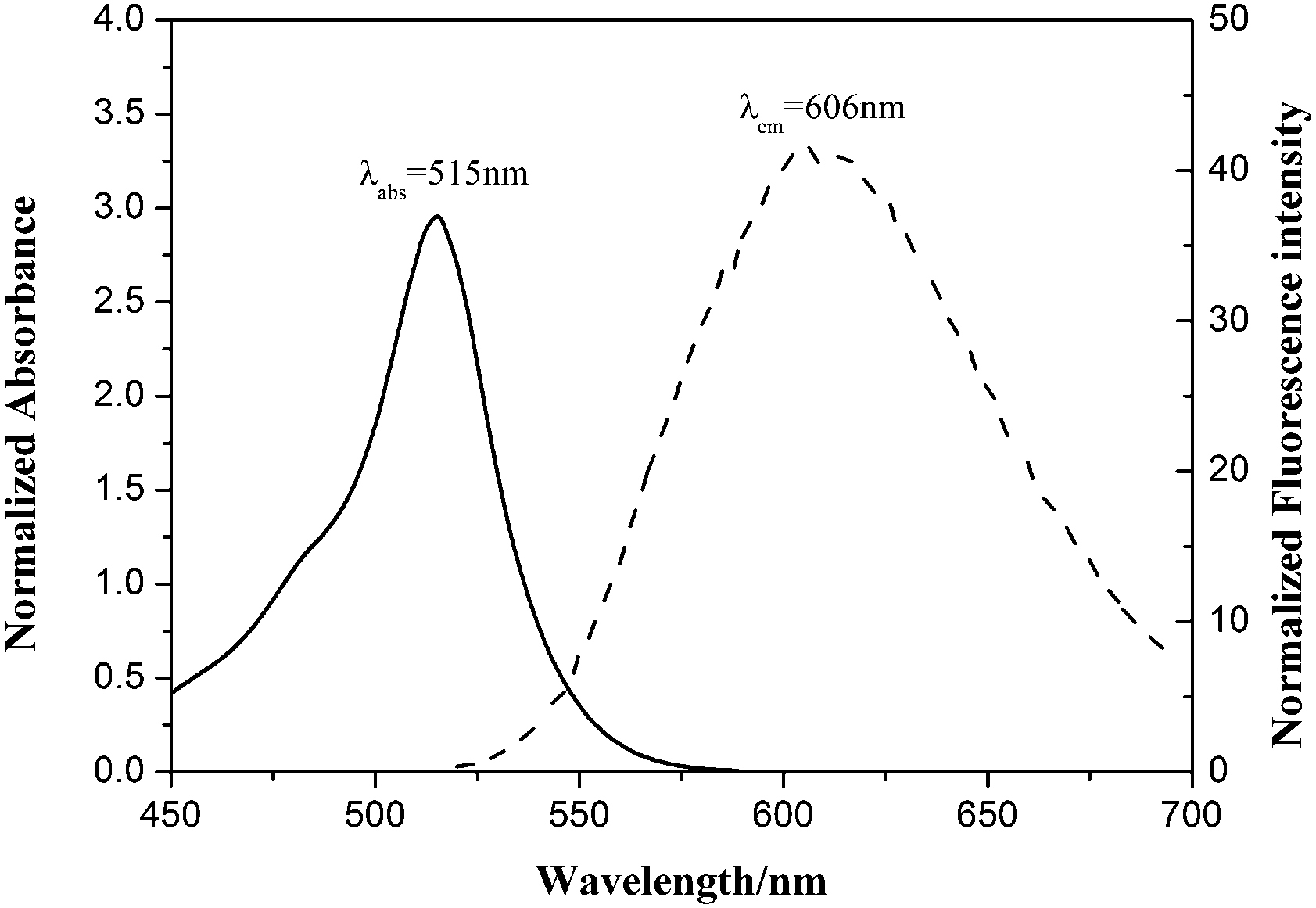 | ||
| Fig. 2 Normalized absorption (solid) and emission (dash) spectra of BODIPY 1 in dichloromethane. | ||
Akkaya et al. also detected the photophysical properties of BODIPY 1 in chloroform, which showed a maximum absorption and emission at 515 and 588 nm, respectively. Its τ and Φ△ were determined to be 10.9 ns (in reference to rhodamine 6G in ethanol) and 0.46 (in reference to methylene blue in CH2Cl2), respectively,11 which is much higher than the reported unhalogenated BODIPYs and many other organic chromophores and photosensitizers under comparable conditions.
The selective oxidation of sulfides to the corresponding sulfoxides is one of the most fundamental organic transformations due to the fact that sulfoxides are important intermediates for various valuable compounds.23 The photocatalytic activities of BODIPY 1 were then evaluated using thioanisole as the substrate (Fig. 3). The reaction was carried out in methanol at room temperature without any additives. A 24 W household fluorescent lamp with a highpass filter (λ = 395 nm) was used as the visible light source (400–700 nm).16 As shown from Fig. 3, BODIPY 1 was highly effective for the oxidation of thioanisole to the corresponding (methylsulfinyl)benzene. The conversion was up to 99% within 6 h and no overoxidation product was detected (Table 1).
 | ||
| Fig. 3 Time effect on the conversion of thioanisole. Reaction conditions: thioanisole (0.5 mmol), MeOH (1 mL), BODIPY 1 (1 mol%), 24 W fluorescent lamp, rt. | ||
| BODIPY | Solvent | λabs (nm) | λem (nm) | Stocks shift (nm) | Φfa |
|---|---|---|---|---|---|
| a Fluorescence quantum yields (Φ) were calculated based on BODIPY 2 in anhydrous ethanol (Φ = 0.98, c = 10 μmol L−1). | |||||
| 1 | Hexane | 514 | 563 | 49 | 0.721 |
| CH2Cl2 | 515 | 606 | 91 | 0.085 | |
| THF | 514 | 605 | 91 | 0.164 | |
| Ethanol | 512 | 610 | 98 | 0.019 | |
| Methanol | 512 | 614 | 102 | 0.004 | |
| 6 | Hexane | 512 | 563 | 51 | 0.046 |
| CH2Cl2 | 513 | 532 | 19 | 0.007 | |
| THF | 512 | 531 | 19 | 0.022 | |
| Ethanol | 510 | 525 | 15 | 0.005 | |
| Methanol | 510 | 522 | 12 | 0.006 | |
| 7 | Hexane | 506 | 535 | 29 | 0.002 |
| CH2Cl2 | 512 | 538 | 28 | 0.009 | |
| THF | 509 | 526 | 17 | 0.024 | |
| Ethanol | 510 | 531 | 21 | 0.008 | |
| Methanol | 509 | 525 | 16 | 0.017 | |
| 8 | Hexane | 511 | 534 | 23 | 0.007 |
| CH2Cl2 | 512 | 533 | 21 | 0.016 | |
| THF | 512 | 536 | 24 | 0.042 | |
| Ethanol | 510 | 529 | 19 | 0.013 | |
| Methanol | 510 | 523 | 13 | 0.001 | |
| 9 | Hexane | 505 | 537 | 32 | 0.006 |
| CH2Cl2 | 512 | 536 | 24 | 0.017 | |
| THF | 507 | 534 | 27 | 0.036 | |
| Ethanol | 510 | 532 | 22 | 0.014 | |
| Methanol | 509 | 522 | 13 | 0.003 | |
| 10 | Hexane | 511 | 540 | 29 | 0.003 |
| CH2Cl2 | 513 | 543 | 30 | 0.012 | |
| THF | 511 | 542 | 31 | 0.030 | |
| Ethanol | 510 | 542 | 32 | 0.009 | |
| Methanol | 510 | 524 | 14 | 0.004 | |
Other parameters on the reaction were also evaluated (Table 2). Among the solvents tested, methanol showed the highest activity. Reactions in non-poplar solvents such as CH2Cl2 and toluene gave trace yields (Table 2, entries 3 and 4). Other BODIPY derivatives 2–10 including either unsubstituted or halogenated BODIPYs exhibited relative lower activities (30–86%, entries 5–13). Further investigations clearly show that other photocatalysts such as rhodamine B and Nile Red were much less effective for the transformation, and only ca. 10% yields were observed. It was worth noting that a 79% yield was obtained after 6 h when Ru(bpy)3Cl2 was utilized, indicating that the photocatalytic activity of BODIPY 1 was higher than the widely used photocatalyst Ru(bpy)3Cl2 in this reaction. Control experiments also showed that no conversion of thioanisole was obtained in the absence of any catalyst or visible light. In addition, trace amount of product was obtained under N2 protection. All these suggested that BODIPY, visible light and oxygen were essential for this reaction (Scheme 3).
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Conversionb (%) |
| a Reaction conditions: thioanisole (0.5 mmol), MeOH (1 mL), catalyst (1 mol%), 24 W fluorescent lamp, rt, 6 h.b Conversion based on NMR. | |||
| 1 | BODIPY 1 | MeOH | 99 |
| 2 | BODIPY 1 | CH3CN | 18 |
| 3 | BODIPY 1 | CH2Cl2 | Trace |
| 4 | BODIPY 1 | Toluene | Trace |
| 5 | BODIPY 2 | MeOH | 42 |
| 6 | BODIPY 3 | MeOH | 65 |
| 7 | BODIPY 4 | MeOH | 53 |
| 8 | BODIPY 5 | MeOH | 86 |
| 9 | BODIPY 6 | MeOH | 33 |
| 10 | BODIPY 7 | MeOH | 41 |
| 11 | BODIPY 8 | MeOH | 32 |
| 12 | BODIPY 9 | MeOH | 37 |
| 13 | BODIPY 10 | MeOH | 30 |
| 14 | Rhodamine B | MeOH | 10 |
| 15 | Nile red | MeOH | 14 |
| 16 | Ru(bpy)3Cl2 | MeOH | 79 |

 | ||
| Scheme 3 Control experiments. | ||
A series of sulfides were tested under the optimized reaction conditions to evaluate the scope and limitations of the current procedure (Table 3). In general, all the reactions proceeded smoothly to give the corresponding products in good yields (82–99%). Sulfides bearing both electron-withdrawing and electron-donating groups showed good activities. The hindrance was also examined and ortho-substituted sulfide gave relative lower yield (Table 3, entry 5). Moreover, no sulfone products were detected in the reactions, demonstrating excellent selectivities of these reactions.

Based on the results and the plausible mechanism proposed by Jing,16,17 the photochemically generated singlet oxygen is the key. It is highly likely that the reaction proceed via the following pathway: first, BODIPY 1 accepted a photon from the visible light to form the excited BODIPY 1*; then the singlet oxygen (1O2)was generated by energy transfer from BODIPY 1* and O2. Alternatively, BODIPY 1* maybe underwent intersystem crossing (ISC) from 1BODIPY 1* to the triplet excited state 3BODIPY 1*, which then reacted with ground state triplet oxygen (3O2) by an energy transfer process, giving singlet oxygen 1O2. Finally, the sulfide was oxidized to form the sulfoxide by singlet oxygen (Scheme 4).
 | ||
| Scheme 4 Proposed mechanism. | ||
In summary, a simple one-pot condensation of 2,4-dimethylpyrrole and oxalyl dichloride to provide an orthogonal dimeric BODIPY 1 has been developed. BODIPY 1 was successfully utilized as a visible-light-driven photocatalyst for the oxidation of sulfides, affording the corresponding sulfoxides in excellent yields and selectivities. In addition, meso-carbalkoxylated BODIPYs, for the first time, were prepared using the similar way via one-pot condensation of 2,4-dimethylpyrrole, oxalyl dichloride and a series of alcohols, which was a good complement for the current BODIPY derivatives. Further investigations on the BODIPY-catalyzed organic reactions are currently underway in our laboratory.
Experimental
General remarks
2,4-dimethylpyrrole, oxalyl dichloride were obtained from Aldrich (Shanghai, China). Other commercially available reagents were used without further purification. 1H- and 13C-NMR spectra were recorded at 500 MHz in CDCl3 using TMS as internal standard Chemical shifts were reported in ppm (δ), and coupling constants (J), in Hz. High resolution mass spectra were determined by EI in a Thermofisher MAT 95 XP. Absorption spectra were performed by using a Varian Cary6000i UV-VIS-NIR absorption spectrophotometer. All the sulfoxides and BODIPYs 2–5 are known compounds and were identified by comparing of their physical and spectra data with those reported in the literature.Typical procedure for one-pot synthesis of BODIPY 1
In N2 bubbled 40 mL dichloromethane, 2,4-dimethylpyrrole (2.05 mL, 20 mmol) and oxalyl dichloride (0.43 mL, 5 mmol) were mixed. The reaction mixture turned red immediately and was kept stirring for 2 h at room temperature. After completion of the reaction, BF3–Et2O (6 mL) was added to the above mixture, followed by dropwise addition of triethylamine (4 mL). After stirring for 3 h at room temperature, the solvent was removed by evaporation under vacuum and a dark residue was obtained which was purified via chromatography on silica gel column, with the eluting solvent of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexane–dichloromethane, giving a red powder (0.51 g, 10%). 1H NMR (500 MHz, CDCl3) δ: 6.03 (s, 4H, ArH), 2.57 (12H, s, CH3), 1.90 (3H, s, CH3); 13C NMR (125 MHz, CDCl3) δ: 157.2, 142.6, 121.6, 14.9, 14.3.
:1 hexane–dichloromethane, giving a red powder (0.51 g, 10%). 1H NMR (500 MHz, CDCl3) δ: 6.03 (s, 4H, ArH), 2.57 (12H, s, CH3), 1.90 (3H, s, CH3); 13C NMR (125 MHz, CDCl3) δ: 157.2, 142.6, 121.6, 14.9, 14.3.
Typical procedure for one-pot synthesis of BODIPY 6–11
In N2 bubbled 40 mL dichloromethane, 2,4-dimethylpyrrole (1 mL, 10 mmol), oxalyl dichloride (0.43 mL, 5 mmol) and an alcohol (5 mmol) were mixed. The reaction mixture turned red immediately and was kept stirring for 1 h at room temperature. After completion of the reaction, BF3–Et2O (6 mL) was added to the above mixture, followed by dropwise addition of triethylamine (4 mL). After stirring for 3 h at room temperature, the solvent was removed by evaporation under vacuum and a dark residue was obtained which was purified via chromatography on silica gel column, with the eluting solvent of 1:1 hexane–dichloromethane, giving a red powder. Other BODIPYs 2–5 were prepared according to the literature.10
Typical procedure for the oxidation of sulfide
To a 10 mL vial equipped with a magnetic stir bar were added BODIPY catalysts (0.05 mmol, 0.01 equiv.), sulfide (0.5 mmol, 1.0 equiv.), and methanol (1 mL). The reaction mixture was stirred at room temperature in air at a distance of ∼5 cm from a 24 W fluorescent lamp with a filter (λ = 395 nm), which was used to emit a small amount of ultraviolet light. 1H NMR spectra was taken of the reaction mixture, and the ratio of integrated intensity between the 1H NMR peaks of the substrate and product was used to calculate the conversion yields.Acknowledgements
This project was financially supported by the National Natural Science Foundation of China (no. 21302014), the Natural Science Foundation for Colleges and Universities of Jiangsu Province, and the Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology (BM2012110).Notes and references
- M. L. Marin, L. S. Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710 CrossRef CAS PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS PubMed.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130 RSC.
- V. Leen, P. Yuan, L. Wang, N. Boens and W. Dehaen, Org. Lett., 2012, 14, 6150 CrossRef CAS PubMed.
- X. Zhang, Y. Xiao, J. Qi, J. Qu, B. Kim, X. Yue and K. D. Belfield, J. Org. Chem., 2013, 78, 9153 CrossRef CAS PubMed.
- H. Qi, J. J. Teesdale, R. C. Pupillo, J. Rosenthal and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 13558 CrossRef CAS PubMed.
- F. Marsico, A. Turshatov, K. Weber and F. R. Wurm, Org. Lett., 2013, 15, 3844 CrossRef CAS PubMed.
- A. B. Nepomnyashchii, A. J. Pistner, A. J. Bard and J. Rosenthal, J. Phys. Chem. C, 2013, 117, 5599 CAS.
- M. Isik, T. Ozdemir, I. S. Turan, S. Kolemen and E. U. Akkaya, Org. Lett., 2013, 15, 216 CrossRef CAS PubMed.
- L. Wang, J. Wang, A. Cui, X. Cai, Y. Wan, Q. Chen, M. He and W. Zhang, RSC Adv., 2013, 3, 9219 RSC.
- Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 123, 12143 CrossRef.
- W. Li, W. Zhang, X. Dong, L. Yan, R. Qi, W. Wang, Z. Xie and X. Jing, J. Mater. Chem., 2012, 22, 17445 RSC.
- A. Kamkaew and K. Burgess, J. Med. Chem., 2013, 56, 7608 CrossRef CAS PubMed.
- A. Vázquez-Romero, N. Kielland, M. J. Arévalo, S. Preciado, R. J. Mellanby, Y. Feng, R. Lavilla and M. Vendrell, J. Am. Chem. Soc., 2013, 135, 16018 CrossRef PubMed.
- N. Adarsh, M. Shanmugasundaram, R. R. Avirah and D. Ramaiah, Chem.–Eur. J., 2012, 18, 12655 CrossRef CAS PubMed.
- W. Li, Z. Xie and X. Jing, Catal. Commun., 2011, 16, 94 CrossRef CAS.
- W. Li, L. Li, H. Xiao, R. Qi, Y. Huang, Z. Xie, X. Jing and H. Zhang, RSC Adv., 2013, 3, 13417 RSC.
- C. Zhang, J. Zhao, S. Wu, Z. Wang, W. Wu, J. Ma, S. Guo and L. Huang, J. Am. Chem. Soc., 2013, 135, 10566 CrossRef CAS PubMed.
- L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627 CrossRef CAS PubMed.
- N. Adarsh, R. R. Avirah and D. Ramaiah, Org. Lett., 2010, 12, 5720–5723 CrossRef CAS PubMed.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162 CrossRef CAS PubMed.
- K. Krumova and G. Cosa, J. Am. Chem. Soc., 2010, 132, 17560 CrossRef CAS PubMed.
- I. Fernandez and N. Khiar, Chem. Rev., 2003, 103, 3651 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and characterization data. See DOI: 10.1039/c4ra01501k |
| This journal is © The Royal Society of Chemistry 2014 |