
Design, synthesis, biological evaluation and molecular docking of novel metronidazole derivatives as selective and potent JAK3 inhibitors
Abstract
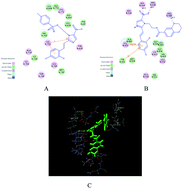
In the JAK/STAT pathway, sustainable activation of JAK with the capabilities of regulating cell growth and apoptosis can produce abnormal proliferation in tumor cells. A series of novel metronidazole derivatives containing the 1,4-benzodioxan moiety as potential inhibitors targeting JAK have been designed, synthesized and their biological activities were also evaluated. Among all synthesized compounds, compound 4t possessed the most potent antitumor activity against A549, Hela, HepG-2 and U251 in vitro, with IC50 values of 65, 21, 16 and 44 nM, respectively, which has been proved by the result of a flow cytometry (FCM) assay. Docking simulations demonstrated that compound 4t could bind tightly with the crystal structure of the JAK3 active site and act as a potential JAK3 inhibitor.
 Please wait while we load your content...
Please wait while we load your content...

