
Deprotonation mechanism of a single-stranded DNA i-motif†
Abstract
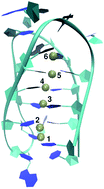
We present the detailed deprotonation mechanism of a fully protonated DNA i-motif as studied by all-atom Molecular Dynamics simulations. The associated unfolding of the i-motif is driven by the application of the Metadynamics method. The release of two protons, which stabilize the hemi-protonated cytosine pairs in the native i-motif, can be identified as the initial step for the occurrence of the unfolding process. By a systematic analysis of the proton motion, we are able to validate a two-step deprotonation mechanism. Our findings are in excellent agreement to experimental results that have been recently published.
 Please wait while we load your content...
Please wait while we load your content...

