A novel fullerene oxide functionalized silica composite as stationary phase for high performance liquid chromatography
Houmei Liuab,
Yong Guoa,
Xusheng Wanga,
Xiaojing Liang*a,
Xia Liu*a and
Shengxiang Jianga
aKey Laboratory for Natural Medicine of Gansu Province, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China. E-mail: gsliuxia@lzb.ac.cn; xjliang@licp.cas.cn; Fax: +86 931 8277088; Tel: +86 931 4968203
bUniversity of the Chinese Academy of Sciences, Beijing 100049, China
First published on 2nd April 2014
Abstract
Hydrophilic interaction liquid chromatography has been widely used for separating hydrophilic compounds and the development of new stationary phases for HILIC is significant. In this study, fullerene oxide was successfully assembled onto silica microspheres to form a FO-modified silica stationary phase. The synthesized material was characterized by elemental analysis, transmission electron microscopy, Raman spectroscopy and contact angle analysis. The chromatographic properties of the stationary phase were investigated in HILIC mode for analysis of nucleosides, nucleobases, water soluble vitamins, amino acids and saccharides. Good separations of these compounds were achieved on the resulting column. Compared with the aminopropylated silica column, FO/SiO2 column exhibited better separation efficiency. This study also investigated the effect of various experimental factors on the retention of the polar stationary phases, such as acetonitrile content and salt concentration in the mobile phase.
1. Introduction
Reversed-phase liquid chromatography (RPLC) is most frequently used in contemporary HPLC practice for separation and purification. However, a major limitation of RPLC lies in its weak retention for hydrophilic compounds. Normal phase liquid chromatography (NPLC), providing a totally different separation mechanism to RPLC, is generally used to separate polar compounds with non-aqueous mobile phases. Nevertheless, hydrophilic compounds are difficult to be dissolved in non-aqueous mobile phases and thus the application of NPLC for separation of hydrophilic compounds is also limited.1 In 1990, the term of hydrophilic-interaction liquid chromatography (HILIC) was first defined by Alpert for the separation of hydrophilic substances such as nucleic acids, proteins, peptides, saccharides and so on.2 In principle, HILIC can be characterized as normal-phase chromatography on polar columns in aqueous–organic mobile phases.3 The retention mechanism of HILIC was originally proposed to be partition. However, due to the complex interactions among the polar stationary phase, mobile phase, the counter-ions of buffer agent and the polar solute, the retention mechanism of HILIC has not been fully established.4 Up to now, stationary phases in HILIC have obtained enormous development.5 Different types of stationary phases for HILIC have their own separation selectivity and retention characteristics. Polar stationary phases in HILIC typically consist of neutral phases (e.g. amide, diol, cross-linked diol),1,7–9 charged phases (e.g. amino, silica),10–14 zwitterionic phases (e.g. sulfobetaine, phosphorylcholine)3,14–16 and other polar stationary phases.17–19 Currently, HILIC has been successfully used for separation of peptides,6,20 carbohydrates,21 drugs,22,23 proteins,24 oligosaccharides,25 metabolites,26 and various natural polar compounds.27–29Carbon materials are important research areas in modern nanoscience, among which fullerene has attracted evergrowing interest. In 1990, Krätschmer and his coworkers successfully obtained macroscopic quantities of fullerene.30 Since then, fullerene has been drawing increasing attention and more and more scientists all over the world are becoming interested in its properties and applications. Fullerene has excellent thermal stability, mechanical and electrical properties, which makes it a candidate stationary phase for chromatography.31–33 However, two main disadvantages make fullerene fail to be direct packing material. As the molecular dimension of normal fullerene material is nanoscale, the permeability will be poor and column pressure will be fairly high when being directly packed in column. The other reason lies in the lack of reactive groups on fullerene, which makes it difficult to be grafted on matrices.34 Therefore, the application of fullerene as direct packing material has been limited to some extent.
The solubility of fullerene in water cannot be high and generally it is incorporated into water-soluble molecules, such as cyclodextrins, to form a “host–guest” complex.30,35 However, as one of the derivatives of fullerene, fullerene oxide (FO) contains a range of reactive oxygen functional groups (e.g. C–O–C, C–OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, COOH) on its surface, which makes it water soluble and enables the covalent incorporation of FO into inorganic or organic matrices.36–38
O, COOH) on its surface, which makes it water soluble and enables the covalent incorporation of FO into inorganic or organic matrices.36–38
In our laboratory, we have already successfully grafted graphene and carbon nano-tube onto silica surface, achieving good separation results.34,39,40 Inspired by the same idea, we assembled FO onto the surface of silica. Owning to the unique properties and structure of FO, this composite will be a promising stationary phase in HILIC.
In this study, FO was immobilized on amino-derivatized silica microparticles and the synthesized material was successfully applied for the separation of hydrotropic substances, including nucleosides, nucleobases, water soluble vitamins, amino acids and saccharides. Compared with the aminopropylated silica, FO-functionalized stationary phase exhibited marvelous separation abilities.
2. Experimental
2.1 Apparatus and reagents
All chromatographic tests were performed on two Agilent 1100 Series modular HPLC systems both with a binary pump, a 20 μL sample loop, and one with a UV-Vis detector, another with a evaporative light-scattering detector. Separations were carried out using columns of 150 mm × 4.6 mm I.D. Deionized water and acetonitrile (analytical grade) were both filtered through a 0.45 μm nylon membrane filter and were degassed ultrasonically prior to use. All samples used in chromatographic tests were analytical-grade reagents.Silica spheres were synthesized using the polymerization-induced colloid aggregation method in our laboratory. The average particle size was 5 μm. The specific surface area and pore diameter were 150 m2 g−1 and 15 nm, respectively. Fullerene with purity over 98% (containing more than 87% C60 and 11% C70), was purchased from Alfa Aesar company (Beijing, China).
2.2 Synthesis of fullerene oxide (FO) functionalized silica stationary phase (FO/SiO2)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V (68% HNO3) = 3:1) with stirring. After 12 h acidification, FO was washed with deionized water and methanol in turn then dried under vacuum for 12 h at 60 °C. 0.2 g of FO was added to 100 mL of deionized water and after the lengthily ultrasonic treatment for 1 h, FO can be dispersed in the deionized water to make 2 mg mL−1 FO dispersion.
:V (68% HNO3) = 3:1) with stirring. After 12 h acidification, FO was washed with deionized water and methanol in turn then dried under vacuum for 12 h at 60 °C. 0.2 g of FO was added to 100 mL of deionized water and after the lengthily ultrasonic treatment for 1 h, FO can be dispersed in the deionized water to make 2 mg mL−1 FO dispersion.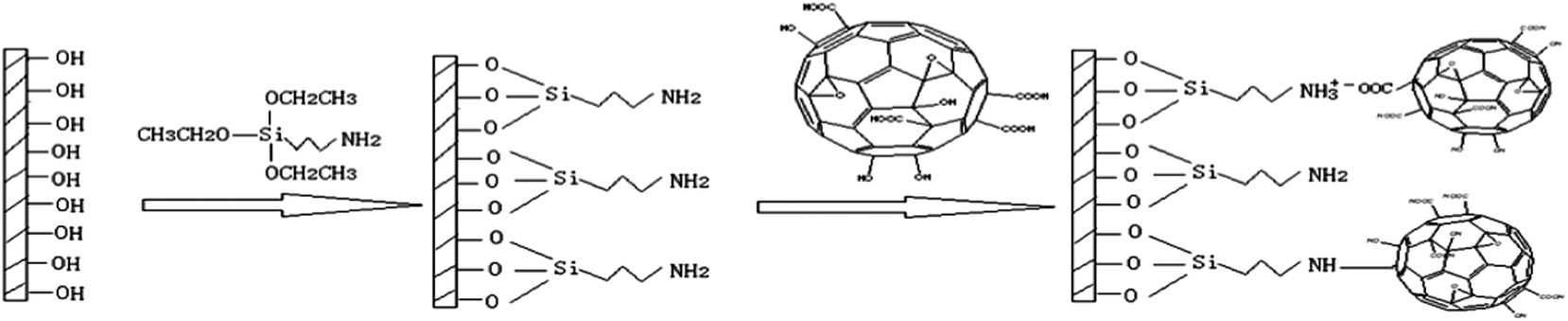 | ||
| Fig. 1 Schematic diagram of preparation of FO functionalized silica. | ||
2.3 Characterization of aminopropylated silica and FO/SiO2 particles
The elemental analyses of aminopropylated silica and FO/SiO2 were performed on a Vario EL (Elementar, Germany). Raman spectrum of FO/SiO2 and aminopropylated silica were performed on inVia-Reflex laser confocal Raman spectrometer (Renishaw, UK). Sessile water-droplet contact angle values were acquired using a DSA-100 optical contact-angle meter (Kruss, Germany) at ambient temperature.2.4 Column packing
Columns (150 × 4.6 mm I.D.) were made of stainless steel tubing and were downward packed using a slurry method with tetrachloromethane as the solvent. A 40 MPa packing press (6752B-100, Beijing, China) was used; hexane was used as the propulsive solvent.2.5 Conditions for chromatographic evaluation
The mixtures of nucleosides and water soluble vitamins were analyzed at room temperature at a flow rate of 1.0 mL min−1 with the ultraviolet (UV) detector at 254 nm and 260 nm, respectively. Amino acids and saccharides were tested with evaporative light scattering detector (ELSD), with the tube temperature at 115.0 °C and gas flow at 2.0 L min−1. Each analyte was dissolved with the mobile phase.3. Results and discussion
3.1 Characterization of aminopropylated silica and FO/SiO2 particles
Elemental analysis data of aminopropylated silica and FO/SiO2 are listed in Table 1. From the carbon content increasing from 2.71% to 3.21%, the surface coverage of FO onto silica was calculated to be 46.3 nmol m−2. The calculation formula of surface coverage of FO is as follow: surface coverage of FO (nmol m−2) = (C% × 107)/(12 × 60 × S). C% represents the percentage of the increased carbon content and S is the specific surface area of SiO2 (150 m2 g−1).| Different particles | Elemental analyses data | ||
|---|---|---|---|
| N (%) | C (%) | H (%) | |
| Aminopropylated silica | 0.76 | 2.71 | 1.14 |
| FO/SiO2 | 0.48 | 3.21 | 1.18 |
Fig. 2 shows the Raman spectrum for the FO/SiO2 and aminopropylated silica in the wave-number range between 100 cm−1 and 3200 cm−1. From Fig. 2, it can be seen that the three dominant and one medium Raman peaks of FO/SiO2 are located at 1468 cm−1, 493 cm−1, 268 cm−1 and 710 cm−1, respectively. However, there are no peaks at these four wave-numbers for aminopropylated silica particles. The four Raman peaks are all characteristic peaks of fullerene.41,42 Consequently, we can confirm that the fullerene was successfully grafted onto the silica.
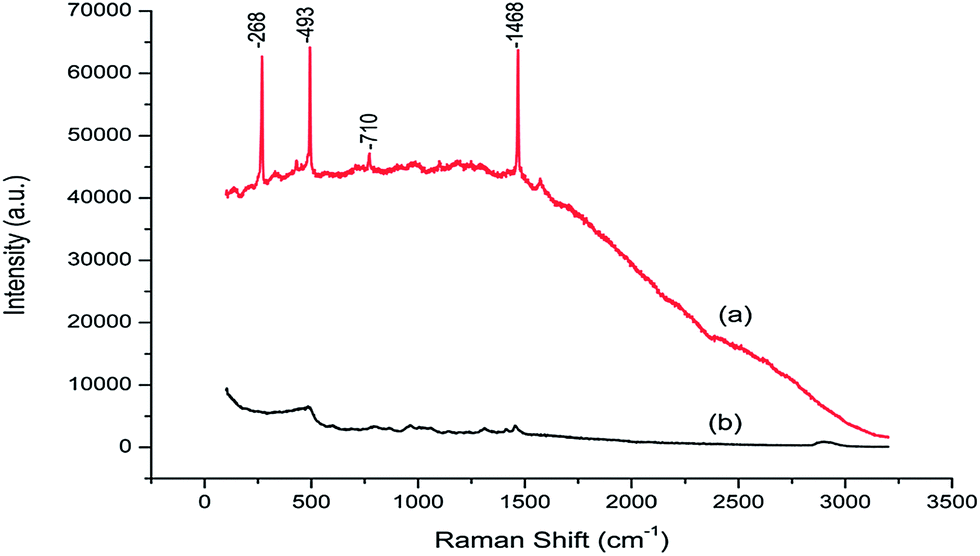 | ||
| Fig. 2 Raman spectrum of FO/SiO2 (a) and aminopropylated silica (b). | ||
We also measured the contact angle of FO/SiO2 and the result was 13.8°, implying that FO/SiO2 is a kind of strong hydrophilic material.
3.2 Chromatographic separation of nucleosides and nucleobases
The level of organic solvent in the mobile phase is probably the most important influence factor on retention. In this study, the effect of acetonitrile content on retention was investigated by varying the percentage of acetonitrile in the mobile phase while keeping ammonium acetate concentration constant at 50 mM. The retention factors of nucleosides and nucleobases were plotted against the acetonitrile content in the mobile phase on aminopropylated silica and FO/SiO2 columns. As shown in Fig. 3, both the two columns exhibited typical HILIC behaviors of increasing retention with increasing acetonitrile content in the mobile phase.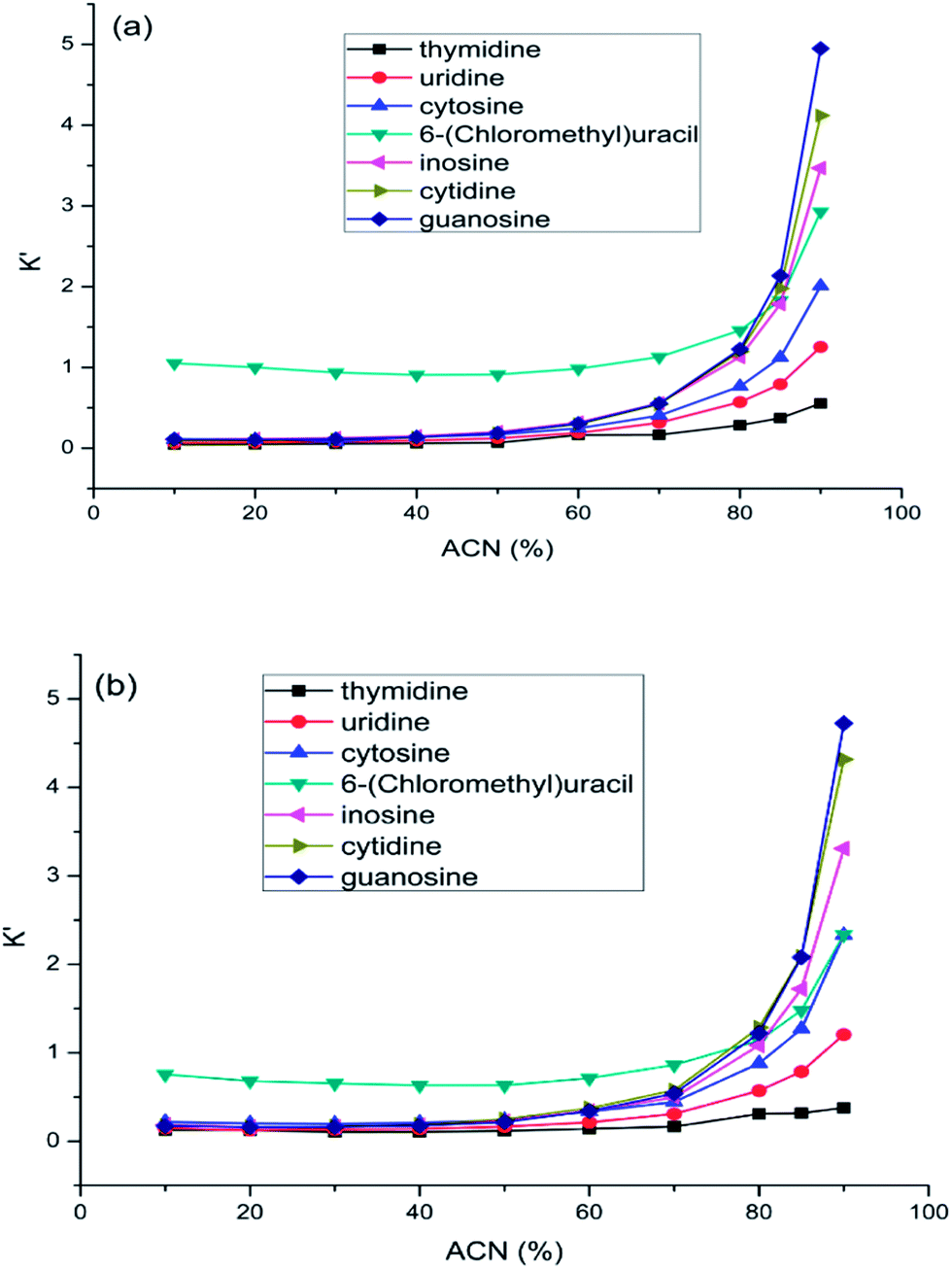 | ||
| Fig. 3 The effect of acetonitrile content on the retention of nucleosides and nucleobases on FO/SiO2 column (a) and aminopropylated silica column (b). Column temperature was room temperature and the mobile phase contained 50 mM ammonium acetate. Flow rate: 1.0 mL min−1. UV detection at 245 nm. | ||
The mixture of nucleosides and nucleobases was separated on the both columns, as shown in Fig. 4. The retention and elution order were the same on the two columns. Exceptionally, cytosine and 6-(chloromethyl)uracil coeluted on the aminopropylated silica column, while all the compounds were well separated on the FO/SiO2 column. Compared with the aminopropylated silica column, FO/SiO2 column exhibited stronger retention for 6-(chloromethyl)uracil, which can be ascribed to the p–π conjugate interaction between unpaired electrons of chlorine and large π system of FO. Meanwhile, we can notice that the peaks were broader for FO/SiO2 than aminopropylated silica column. Because the particle size of FO is on the same order of magnitude with the pore path size on SiO2 surface, the FO bonded onto SiO2 would inevitably damage the original uniform holes, which caused the efficiency decrease of FO/SiO2 column.
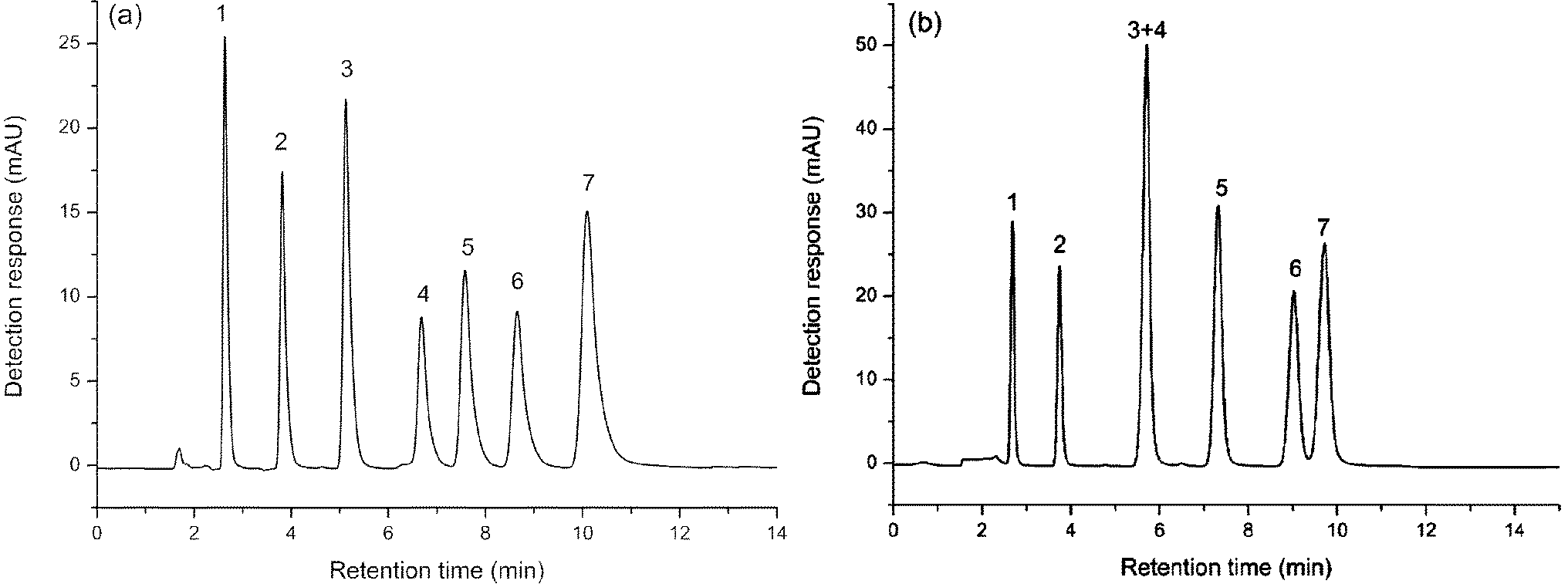 | ||
| Fig. 4 Separation of nucleosides and nucleobases on: FO/SiO2 column (a) and aminopropylated silica column (b). Mobile phase: acetonitrile–water (90/10, v/v) containing 50 mM ammonium acetate. Column temperature: room temperature. Flow rate: 1.0 mL min−1. UV detection at 245 nm. Compounds: (1) thymidine, (2) uridine, (3) cytosine, (4) 6-(chloromethyl)uracil, (5) inosine, (6) cytidine and (7) guanosine. | ||
3.3 Chromatographic separation of water soluble vitamins
The buffer concentration effect on separation of water soluble vitamins was investigated on both aminopropylated silica and FO/SiO2 columns (Fig. 5). As can be seen in Fig. 5(a), that the retention of water soluble vitamins almost had no change on FO/SiO2 column. However, the retentions of vitamin B3 and vitamin C decreased, while vitamin B1 increased, with the increasing of buffer concentration on aminopropylated silica column. This demonstrated that ion exchange mechanism existed on aminopropylated silica column, while not on FO/SiO2 column. For vitamin B3 and vitamin C, the electrostatic attraction interaction was suppressed by increasing buffer concentration and then the retention decreased, and it was opposite for vitamin B1. This phenomenon also illustrated that the mixed-mode feature on aminopropylated silica column and HILIC mode on FO/SiO2 column.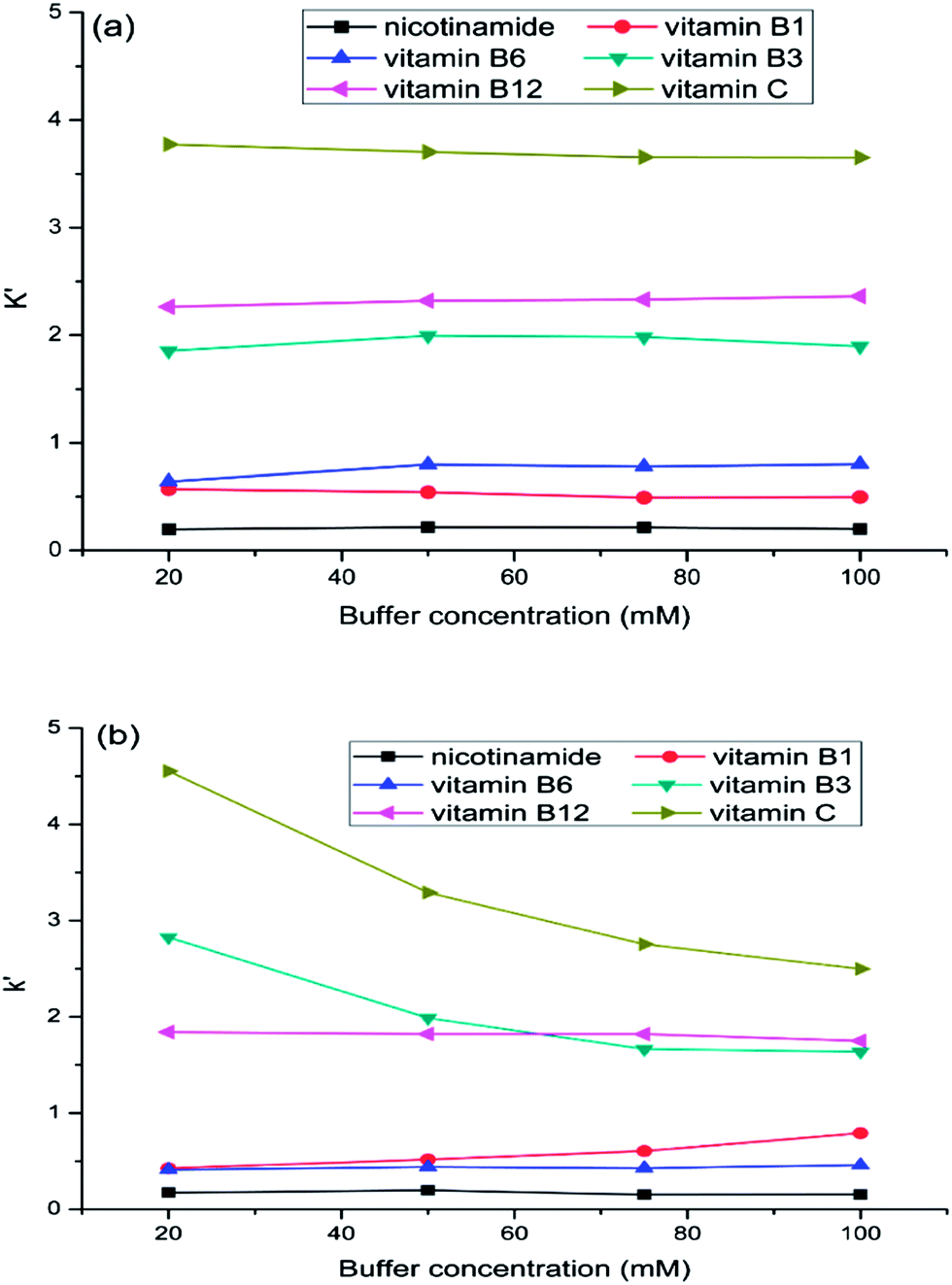 | ||
| Fig. 5 The effect of buffer concentration on water soluble vitamins separation: FO/SiO2 column (a) and aminopropylated silica column (b). Mobile phase: acetonitrile–water (90/10, v/v) containing 20, 50, 75, 100 mM ammonium acetate, respectively. Column temperature: room temperature. Flow rate: 1.0 mL min−1. UV detection at 260 nm. | ||
As shown in Fig. 6(a), baseline separation of six water soluble vitamins could be achieved under the optimal conditions on FO/SiO2 column. However, vitamin B1 and vitamin B6, vitamin B3 and vitamin B12 were failed to be totally separated on aminopropylated silica column. The elution order of vitamin B1 and vitamin B6 was inversed on the two columns. Due to the existence of relatively stronger hydrophilic interaction between vitamin B6 and the FO/SiO2 stationary phase, vitamin B6 had longer retention time compared to the retention on aminopropylated silica column. It was also found that the peak of vitamin B12 was leading peak. Because the vitamin B12 is a class of water soluble vitamin containing Co1+, there is charge repulsion between vitamin B12 and aminopropylated silica stationary phase. Consequently, under the resultant forces of hydrophilic retention and charge repulsion, the peak of vitamin B12 was leading.
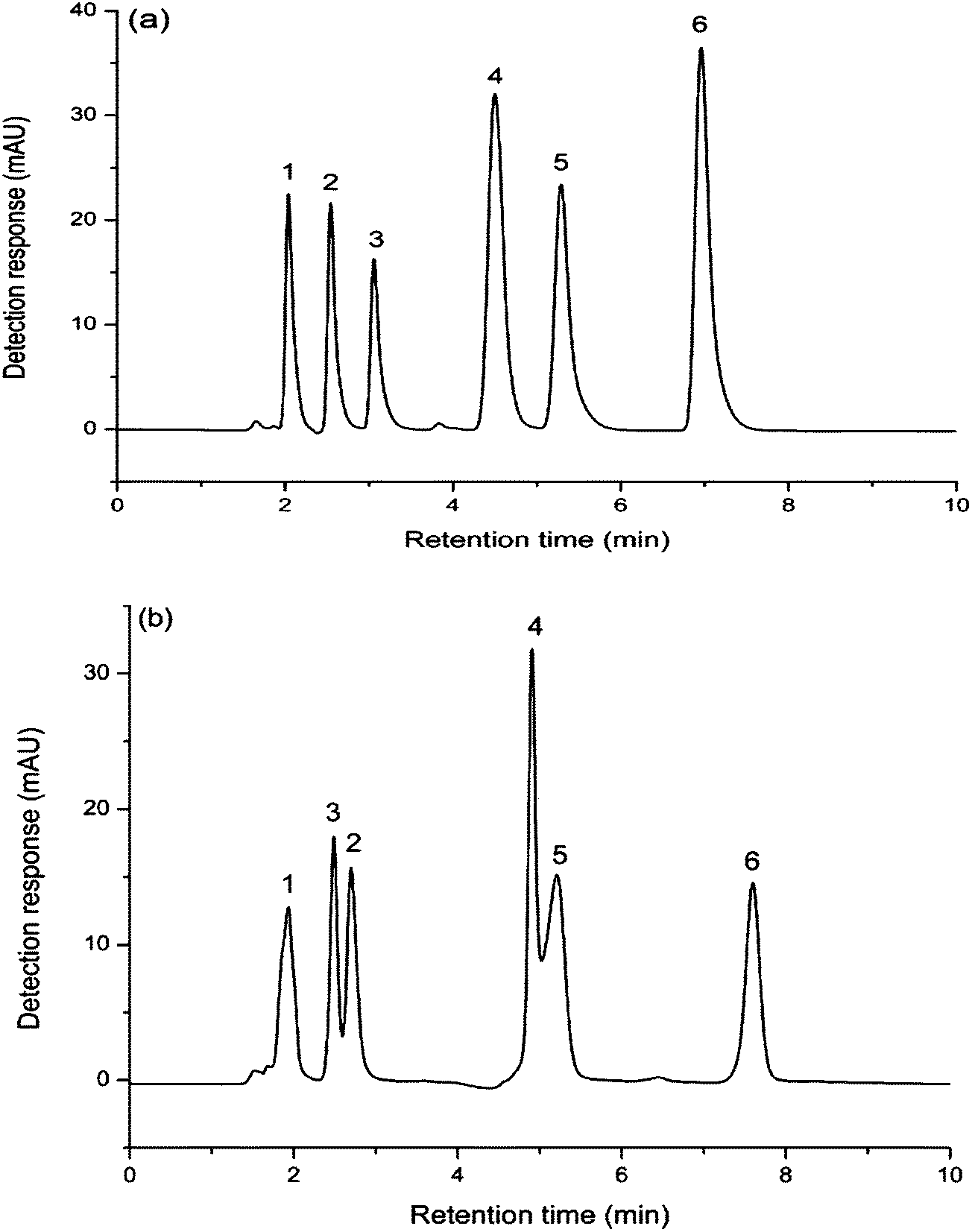 | ||
| Fig. 6 Separation of water soluble vitamins on: FO/SiO2 column (a) and aminopropylated silica column (b). Mobile phase: acetonitrile–water (73/27, v/v) containing 100 mM ammonium acetate. Column temperature: room temperature. Flow rate: 1.0 mL min−1. UV detection at 260 nm. Compounds: (1) nicotinamide, (2) vitamin B1, (3) vitamin B6, (4) vitamin B3, (5) vitamin B12 and (6) vitamin C. | ||
3.4 Chromatographic separation of amino acids
A test mixture of DL-phenylalanine, DL-methionine, DL-valine, L-proline, L-serine, L-arginine was investigated on these two columns with a mobile phase of acetonitrile–water (70/30, v/v) containing 50 mM ammonium acetate, and the separation chromatograms are shown in Fig. 7.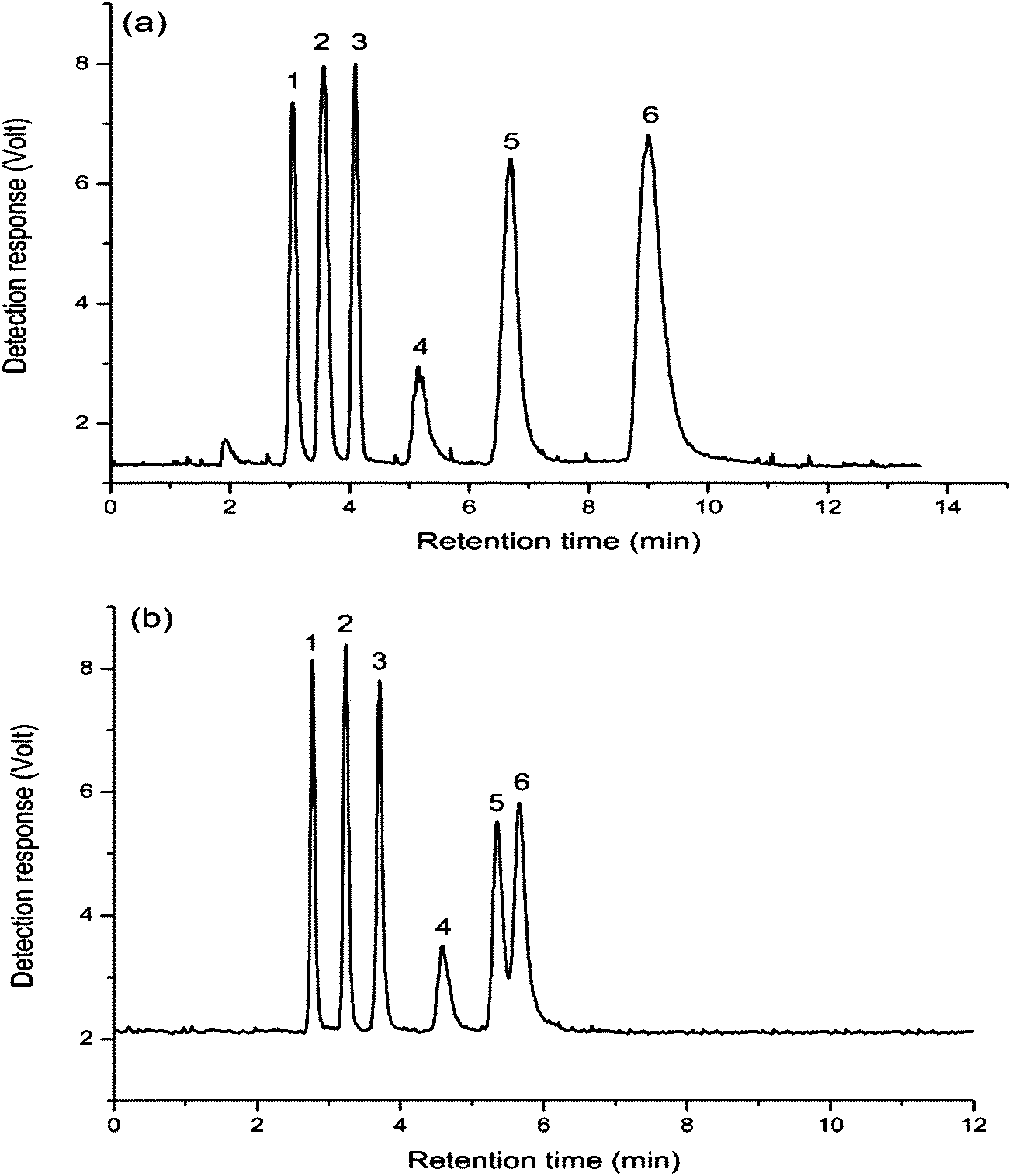 | ||
| Fig. 7 Separation of amino acids on: FO/SiO2 column (a) and aminopropylated silica column (b). Mobile phase: acetonitrile–water (70/30, v/v) containing 50 mM ammonium acetate. Column temperature: room temperature. Flow rate: 1.0 mL min−1. ELS detector: gas flow: 2 L min−1, tube temperature 115 °C. Compounds: (1) DL-phenylalanine, (2) DL-methionine, (3) DL-valine, (4) L-proline, (5) L-serine and (6) L-arginine. | ||
Fig. 7 demonstrated that the eluting orders of these amino acids compounds on the two columns were the same. In comparison, all the amino acids showed stronger retentions on the FO/SiO2 column, especially for L-arginine, which indicated that the hydrophilcity of FO-modified stationary phase was stronger than that of aminopropylated silica stationary phase. So six amino acids all achieved baseline separation on FO/SiO2 column, while L-serine and L-arginine were only partially resolved on aminopropylated silica column. As for the phenomenon about broader peaks for FO/SiO2 than aminopropylated silica column, the same reason was already mentioned in the Section 3.2.
3.5 Chromatographic separation of saccharides
Fig. 8 shows the separation of five saccharides compounds including L-rhamnose, DL-arabinose, D-glucose, sucrose, lactose on FO/SiO2 column (a) and aminopropylated silica column (b) with a mobile phase of acetonitrile–water (73/27, v/v) containing 50 mM ammonium acetate as the mobile phase. From the Fig. 8, we can see that the five saccharide compounds could be completely separated on FO/SiO2 column, while the three monosaccharide, L-rhamnose, DL-arabinose and D-glucose, could not be totally separated on aminopropylated silica column, and all peaks on aminopropylated silica column were relatively broad, illustrating that this five saccharides had weak interaction with aminopropylated silica stationary phase, and when the retention was enhanced merely through decreasing the elution power of mobile phase, the peaks inevitably became wider.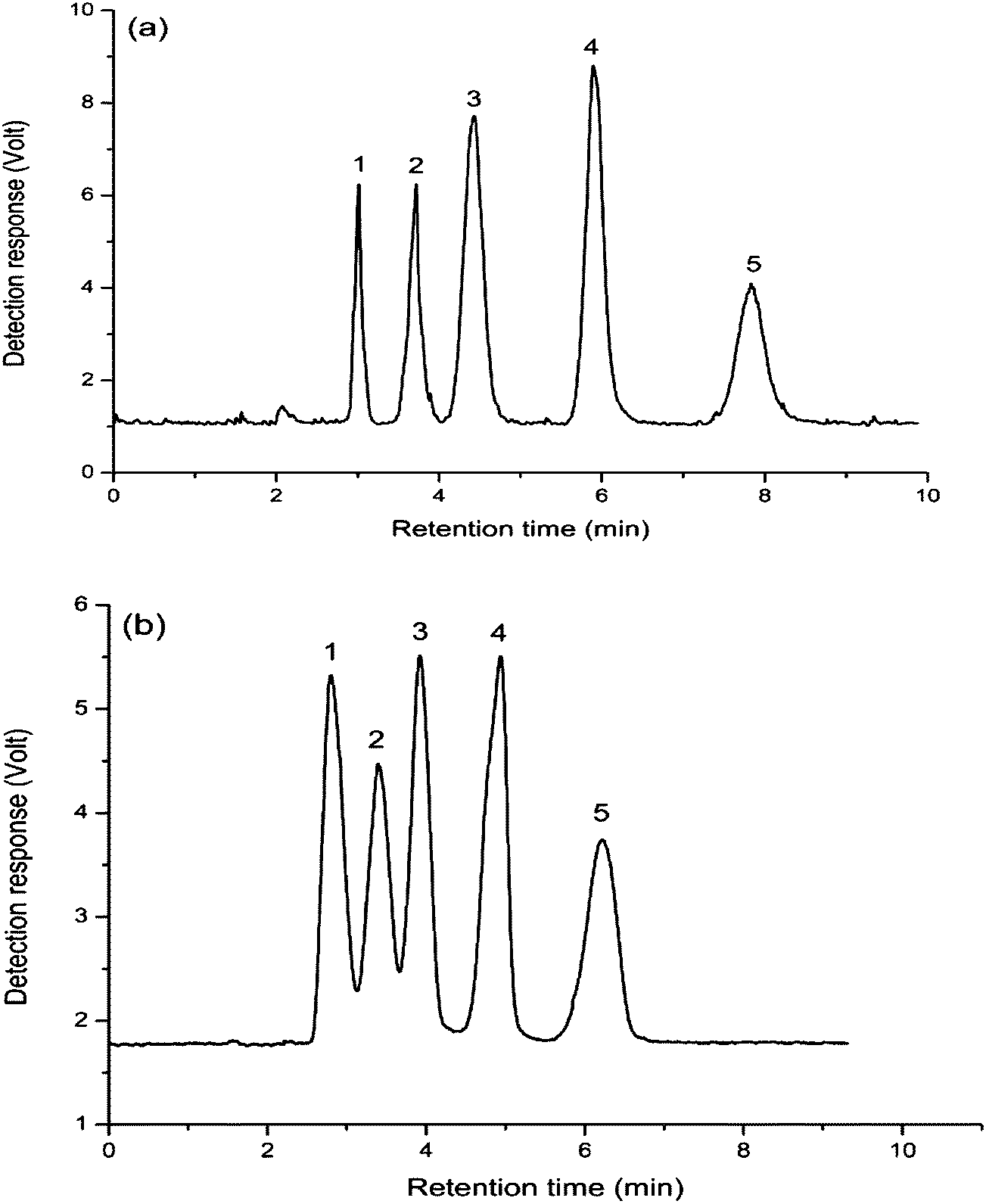 | ||
| Fig. 8 Separation of saccharides on: FO/SiO2 column (a) and aminopropylated silica column (b). Mobile phase: acetonitrile–water (73/27, v/v) containing 50 mM ammonium acetate. Column temperature: room temperature. Flow rate: 1.0 mL min−1. ELS detector: gas flow: 2 L min−1, tube temperature 115 °C. Compounds: (1) L-rhamnose, (2) DL-arabinose, (3) D-glucose, (4) sucrose and (5) lactose. | ||
4. Concluding remarks
Fullerene was oxidized and subsequently successfully bonded onto the surface of silica particles to prepare a HILIC stationary phase (FO/SiO2). The resulting stationary phase displayed excellent selectivity and efficient retention for various polar compounds. The comparison of chromatographic performances of FO/SiO2 column and aminopropylated silica column clearly showed that the former was more hydrophilic and had better separation ability for hydrophilic compounds. The study on the effect of buffer salt concentration on retention provided experimental evidences that the ion-exchange effect was responsible for the retention of charged compounds on the amino-modified phase, but not significantly affected the retention on FO-modified stationary phase. All the results indicated that FO was a novel hydrophilic material and FO-modified stationary phase had its unique application in HILIC. Due to the superiorities of FO, more applications will be further explored in analytical area.Acknowledgements
Financial supports from the National Natural Science Foundation of China (21105107, 21175143 and 20905072) are gratefully acknowledged.References
- T. Yoshida, J. Biochem. Biophys. Methods, 2004, 60, 265 CrossRef CAS PubMed.
- A. J. Alpert, J. Chromatogr., 1990, 499, 177 CrossRef CAS.
- P. Jandera, Anal. Chim. Acta, 2011, 692, 1 CrossRef CAS PubMed.
- P. Hemström and K. Irgum, J. Sep. Sci., 2006, 29, 1784 CrossRef.
- H. D. Qiu, X. J. Liang, M. Sun and S. X. Jiang, Anal. Bioanal. Chem., 2011, 399, 3307 CrossRef CAS PubMed.
- B. Buszewski and S. Noga, Anal. Bioanal. Chem., 2012, 402, 231 CrossRef CAS PubMed.
- Y. Guo and S. Gaiki, J. Chromatogr. A, 2011, 1218, 5920 CrossRef CAS PubMed.
- P. Jandera and T. Hájek, J. Sep. Sci., 2009, 32, 3603 CrossRef CAS PubMed.
- G. J. Patti, J. Sep. Sci., 2011, 34, 3460 CrossRef CAS PubMed.
- V. Pucci, C. Giuliano, R. Zhang, K. A. Koeplinger, J. F. Leone, E. Monteagudo and F. Bonelli, J. Sep. Sci., 2009, 32, 1275 CrossRef CAS PubMed.
- N. S. Quiming, N. L. Denola, Y. Saito and K. Jinno, J. Sep. Sci., 2008, 31, 1550 CrossRef CAS PubMed.
- V. V. Tolstikov and O. Fiehn, Anal. Biochem., 2002, 301, 298 CrossRef CAS PubMed.
- U. Woelwer-Rieck, C. Lankes, A. Wawrzun and M. Wüst, Eur. Food Res. Technol., 2010, 231, 581 CrossRef CAS.
- S. Di Palma, P. J. Boersema, A. J. Heck and S. Mohammed, Anal. Chem., 2011, 83, 3440 CrossRef CAS PubMed.
- G. Greco, S. Grosse and T. Letzel, J. Chromatogr. A, 2012, 1235, 60 CrossRef CAS PubMed.
- D. V. McCalley, J. Chromatogr. A, 2010, 1217, 3408 CrossRef CAS PubMed.
- P. Dallet, L. Labat, E. Kummer and J. Dubost, J. Chromatogr. B: Biomed. Sci. Appl., 2000, 742, 447 CrossRef CAS.
- J.-t. Feng, Z.-m. Guo, H. Shi, J.-p. Gu, Y. Jin and X.-m. Liang, Talanta, 2010, 81, 1870 CrossRef CAS PubMed.
- H. Tanaka, X. Zhou and O. Masayoshi, J. Chromatogr. A, 2003, 987, 119 CrossRef CAS.
- A. R. Oyler, B. L. Armstrong, J. Y. Cha, M. X. Zhou, Q. Yang, R. I. Robinson, R. Dunphy and D. J. Burinsky, J. Chromatogr. A, 1996, 724, 378 CrossRef CAS.
- A. J. Alpert, M. Shukla, A. K. Shukla, L. R. Zieske, S. W. Yuen, M. A. Ferguson, A. Mehlert, M. Pauly and R. Orlando, J. Chromatogr. A, 1994, 676, 191 CrossRef CAS.
- R. Li and J. Huang, Prog. Chem., 2006, 18, 1508 CAS.
- M. A. Strege, S. Stevenson and S. M. Lawrence, Anal. Chem., 2000, 72, 4629 CrossRef CAS.
- P. J. Boersema, N. Divecha, A. J. Heck and S. Mohammed, J. Proteome Res., 2007, 6, 937 CrossRef CAS PubMed.
- G. O. Staples, M. J. Bowman, C. E. Costello, A. M. Hitchcock, J. M. Lau, N. Leymarie, C. Miller, H. Naimy, X. Shi and J. Zaia, Proteomics, 2009, 9, 686 CrossRef CAS PubMed.
- Y. Hsieh, J. Sep. Sci., 2008, 31, 1481 CrossRef CAS PubMed.
- A. M. Ares and J. Bernal, Cent. Eur. J. Chem., 2012, 10, 534 CrossRef PubMed.
- L. Nováková and H. Vlčková, Anal. Chim. Acta, 2009, 656, 8 CrossRef PubMed.
- J. Bernal, A. M. Ares, J. Pól and S. K. Wiedmer, J. Chromatogr. A, 2011, 1218, 7438 CrossRef CAS PubMed.
- W. Krätschmer, L. D. Lamb, K. Fostiropoulos and D. R. Huffman, Nature, 1990, 347, 354 CrossRef.
- L. Kartsova and A. Makarov, Russ. J. Appl. Chem., 2002, 75, 1725 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC.
- M. Fischnaller, R. Bakry, R. M. Vallant, L. A. Huber and G. K. Bonn, Anal. Chim. Acta, 2013, 761, 92 CrossRef CAS PubMed.
- X. Liang, S. Wang, S. Liu, X. Liu and S. Jiang, J. Sep. Sci., 2012, 35, 2003 CrossRef CAS PubMed.
- R. Taylor and D. R. Walton, Nature, 1993, 363, 354 Search PubMed.
- C. A. Mirkin and W. Brett Caldwell, Tetrahedron, 1996, 52, 5113 CrossRef CAS.
- Y. Saito, H. Ohta, H. Terasaki, Y. Katoh, H. Nagashima, K. Jinno, K. Itoh, R. D. Trengove, J. Harrowfield and S. F. Li, J. High Resolut. Chromatogr., 1996, 19, 475 CrossRef CAS.
- D. Stalling, C. Guo and S. Saim, J. Chromatogr. Sci., 1993, 31, 265 CAS.
- X. Liang, S. Liu, H. Liu, X. Liu and S. Jiang, J. Sep. Sci., 2010, 33, 3304 CrossRef CAS PubMed.
- X. Liang, S. Liu, X. Song, Y. Zhu and S. Jiang, Analyst, 2012, 137, 5237 RSC.
- A. A. Arie, J. O. Song and J. K. Lee, Mater. Chem. Phys., 2009, 113, 249 CrossRef CAS PubMed.
- R. L. Garrell, T. M. Herne, C. A. Szafranski, F. Diederich, F. Ettl and R. L. Whetten, J. Am. Chem. Soc., 1991, 113, 6302 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |