Synthesis of multifunctional lipid–polymer conjugates: application to the elaboration of bright far-red fluorescent lipid probes†
Salim Adjiliab,
Arnaud Favier*ab,
Julien Massinc,
Yann Bretonnièrec,
William Lacourab,
Yi-Chun Linab,
Elodie Chatrea,
Christophe Placea,
Cyril Favardd,
Delphine Muriaux‡
e,
Chantal Andraudc and
Marie-Thérèse Charreyre*ab
aÉcole Normale Supérieure de Lyon, Laboratoire Joliot-Curie, CNRS USR 3010, F-69364 Lyon, France. E-mail: arnaud.favier@ens-lyon.fr; marie-therese.charreyre@ens-lyon.fr
bINSA-Lyon, Laboratoire Ingénierie des Matériaux Polymères, CNRS UMR 5223, F-69621 Villeurbanne, France
cÉcole Normale Supérieure de Lyon, Laboratoire de Chimie, CNRS UMR 5182, Université Lyon 1, Site Monod, 46 allée d'Italie, F-69364, Lyon, France
dCentre d'études d'agents Pathogènes et Biotechnologies pour la Santé, CNRS UMR 5236, F-34293, Montpellier, France
eÉcole Normale Supérieure de Lyon, Laboratoire de Virologie Humaine, INSERM U758, F-69364 Lyon, France
First published on 17th March 2014
Abstract
A new class of lipid-ended polymer conjugates presenting reactive sites regularly distributed along the polymer chain were synthesized using RAFT polymerization. The chosen modular approach enables preparation of different lipid families by tuning the nature of the phospholipid α-end, the molecular weight and the lateral functions of the polymer chain. The multiple activated ester functions of the conjugates can indeed be used for the efficient coupling of a great variety of amino-containing entities of interest. In this study, we elaborated original fluorescent lipid–polymer probes for optical microscopy by coupling along the chain a controlled number of chromophores emitting in the far-red where auto-fluorescence and light absorption by biological samples are limited. Water-soluble fluorescent lipid probes exhibiting an enhanced brightness were obtained. As a proof of concept, these probes were able to efficiently label the lipid bilayer of liposomes of various sizes. Such multifunctional lipid-ended polymers thus exhibit great potential to functionalize model and natural lipid assemblies.
Introduction
Lipid–polymer conjugates are increasingly used in a wide range of (bio)applications, the major one being the development of nanomedicine delivery devices.1 Hydrophilic polymer chains carrying a lipid at one chain-end (lipid-ended polymers, LEPs) are very useful to stabilize lipid self-assemblies such as supported bilayers and liposomes. The terminal lipid residue is anchored in the lipid bilayer while the polymer chain provides steric stabilization. In addition, the LEPs may also carry another entity of interest, generally located at the other chain-end, in order to display specific functionalities at the surface of the self-assemblies (e.g. bioactive molecules to improve targeting properties).2–4PEGylated lipids, that are composed of a linear polyethylene glycol (PEG) – a hydrophilic, flexible and inert polymer – covalently bound to the polar head of a lipid, are by far the main family of LEPs encountered in the literature. PEGylated lipids improve the stability of liposomes which, when injected in vivo, show prolonged blood circulation and stealth properties (i.e. limited opsonization).5–9 Moreover, a wide range of chemical functions can advantageously be introduced at the remaining lipid–PEG chain-end,10–12 for instance to enable the coupling of a fluorophore.13,14 However, lateral functionalization of PEG chains still remains a challenge and it is yet difficult to introduce various functions along the chain.
On the other hand, α-, ω- and α,ω-functionalized polymers, can now be synthesized through controlled radical polymerization (CRP) techniques.15–18 However, very few articles report the synthesis of LEPs and they are exclusively dealing with homopolymers.19–23 Among the various CRP techniques, RAFT polymerization is one of the most versatile for the development of (bio)conjugates.24 Our group recently reported the synthesis of a lipid-functionalized RAFT agent (lipid-CTA) that efficiently controlled the homopolymerization of an acrylamide derivative, N-acryloylmorpholine (NAM),22 leading to polymers exhibiting similar properties compared to PEG. Such RAFT homopolymerization resulted in well-defined lipid–P(NAM) conjugates with a controlled molecular weight (MW), a narrow MW distribution and, as confirmed by MALDI-ToF mass spectrometry, an intact lipid α-chain-end. In addition, these lipid–polymer conjugates were successfully used to stabilize LipoParticles assemblies in relatively high ionic strength aqueous solutions.
Yet, no LEPs carrying multiple lateral functionalities along the polymer chain (multifunctional LEP conjugates) were reported to date. However, such structures are highly desirable for numerous applications, since they would enable the coupling of various densities/types of entities and consequently to finely tune the properties of the lipid–polymer conjugates. In the present study, we designed modular LEPs exhibiting (i) a well-defined structure (molecular weight, composition, microstructure and functionality), (ii) a lipid of interest attached at the α-end of an hydrophilic backbone and (iii) multiple reactive functions (Fig. 1, top) regularly distributed along the polymer chain that are further used for the covalent coupling of a controlled number of entities of interest.
 | ||
| Fig. 1 Schematic representation of the multifunctional reactive lipid-ended polymers, LEPs, (top) that were used for the elaboration of fluorescent lipid–polymer probes (bottom, left) labeled with chromophore 1 (bottom, right). (NAM units – green, NAS units – black, F – reactive lateral functions, fluorophores – red stars). | ||
RAFT polymerization was used to synthesize LEPs bearing numerous activated ester lateral functions, well-known for their ability to efficiently bind a large variety of amino-containing (bio)molecules.25,26 The reactive lipid–polymer backbone was synthesized in the presence of a lipid-CTA, by copolymerization of NAM with N-acryloxysuccinimide (NAS), a comonomer pair leading to an excellent control over the architecture of the P(NAM-stat-NAS) chains in terms of molecular weight (MW), dispersity but also composition and microstructure.27 Indeed, we previously showed that RAFT copolymerization of NAM and NAS exhibits an azeotropic composition (NAM/NAS 60/40 mol%) for which there is no compositional drift throughout the polymerization (the co-monomer conversion kinetics are identical). Not only is the microstructure identical from one chain to another – which is inherent to a living copolymerization – but the activated ester units are regularly spaced along the polymer chain.
Among the numerous potential applications of this multifunctional LEP platform, one particular interest in the field of bioimaging is the development of bright fluorescent lipid probes. Fluorescent lipids are used to label natural and/or artificial lipid bilayers such as liposomes.28 For the synthesis of fluorescent lipids, hydrophobic fluorophores are most of the time introduced as part of the fatty acid chains. However, since the bulky and rigid structure of the fluorophore (compared to the natural fatty acids) may alter the insertion properties of the fluorescent lipid into the bilayers, introduction of the fluorophore on the polar head of the lipid may be preferred. In the latter case, a more hydrophilic fluorophore is either directly attached to the polar head or via a PEG linker.13,29–31
Conventional fluorescent lipids, including those commercially available, generally bear only one fluorophore leading to a moderate brightness. Our objective here was to develop fluorescent lipid–polymer probes bearing multiple fluorophores, thus exhibiting an enhanced brightness. The multifunctional LEP platform was therefore used for the covalent coupling of a controlled number of fluorophores along the hydrophilic polymer backbone (Fig. 1, bottom). We selected a push–pull dipolar fluorophore (Fig. 1) emitting in the far-red with interesting two-photon absorption properties. This class of chromophores (derivatives of isophorone) indeed exhibits many advantages for bioimaging applications since, at these operating wavelengths, light absorption and scattering by the biological tissues are low. As a consequence, phototoxicity and auto-fluorescence are reduced, while light penetration into tissues is higher.32,33 This chromophore has recently been used for the elaboration of a water-soluble probe for cerebral vascular imaging.34 It also presents an intense fluorescence in the aggregated state:35 its fluorescence should then be less sensitive to the self-quenching phenomenon expected when multiple fluorophores are bound to the same polymer backbone.
As a proof of concept, several multifunctional LEP conjugates carrying a dipalmitoyl phospholipid at their α-chain-end and exhibiting various chain lengths were synthesized. Then, covalent coupling of the fluorophores along the chain was carried out, resulting in fluorescent LEP conjugates. In addition, the modularity of the multifunctional LEP platform was also used to introduce electrostatic charges that are known to influence the behavior of polymers under bio-relevant conditions. A library of neutral and negatively charged fluorescent LEP conjugates was prepared by varying the polymer molecular weight and the number of fluorophores per polymer chain. After purification, the photophysical properties of the conjugates were investigated using UV-Vis and fluorescence spectroscopies. Finally, optical microscopy was used to assess the ability of the new lipid–polymer probes to label model lipid bi-layers such as liposomes.
Results and discussion
Synthesis and characterization of well-defined multifunctional LEPs
In a first step, lipid-CTAs were synthesized (Scheme 1) from two dipalmitoyl phospholipids carrying a primary amino group, following the general strategy previously described.22 Both lipid-CTAs, A and B (Fig. 2), are derivatives of 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) but the newly synthesized one, B, differed from A by the absence of the C-6 spacer between the dithiobenzoate group and the phosphate of the lipid polar head. The structure of B was confirmed by both 1H NMR spectroscopy (Fig. S1 in ESI†) and mass spectrometry analyses. | ||
| Scheme 1 General synthesis of the lipid-CTAs. | ||
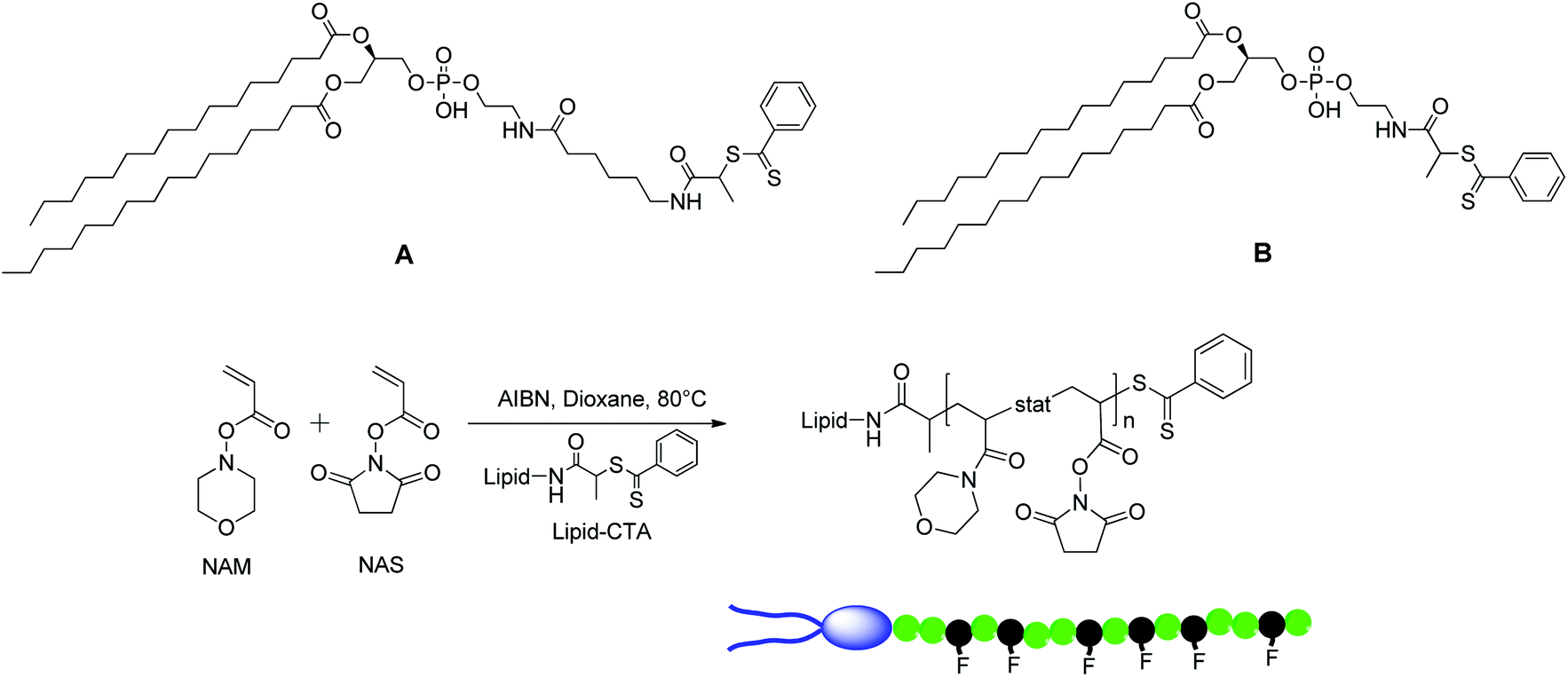 | ||
| Fig. 2 The two lipid-CTAs based on DPPE used in this study (top) to control the RAFT copolymerization of NAM and NAS (bottom). | ||
It has to be noted that purification was improved at various levels compared to the previously described procedure (see also Experimental section): (i) purification of the CAEDB derivative was facilitated by introducing an extraction step to isolate the product before silica gel chromatography which was then easier to conduct and more efficient. (ii) Purity of the SEDB RAFT agent precursor was enhanced by a re-crystallization procedure, that led to a pink powder with a very high purity (>95%). This powder was much easier to handle than the oil generally obtained without re-crystallization. (iii) Such SEDB purity facilitated the synthesis and the purification of the amphiphilic lipid-CTAs. For the latter purification, the silica gel chromatography was advantageously replaced by a silica gel filtration in order to remove the residual salts arising from the extraction procedure. Finally, the two lipid-CTAs were obtained with a 90% yield (>90% purity) relative to the initial phospholipid.
The lipid-CTAs were then used as control agents for the RAFT copolymerization of NAM and NAS. No influence of the presence of NAS in the co-monomer mixture and of the nature of the lipid-CTA was noticed on both polymerization kinetics and MW control (Fig. S2†) in comparison with the good results already reported for NAM homopolymerization.22 In addition, at the 60/40 NAM/NAS initial molar composition, both monomers copolymerized at the same rate (Table 1), confirming that the copolymerization proceeded at the azeotropic composition. Then, the resulting lipid–P(NAM-stat-NAS) copolymers of increasing molecular weight displayed the same composition and microstructure.
| Samplea | NAM conversion (%) | NAS conversion (%) | Mn (g mol−1) | nNASb | Đ |
|---|---|---|---|---|---|
| a The letter A or B refers to the type of lipid-CTA that was used to control the copolymer synthesis.b nNAS is the average number of NAS units per polymer chain. | |||||
| A1 | 26 | 26 | 7900 | 18.1 | 1.06 |
| A2 | 59 | 59 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
50.6 | 1.04 |
| A3 | 85 | 85 | 33200 |
84.5 | 1.11 |
| B1 | 10 | 10 | 5900 | 13.1 | 1.01 |
| B2 | 83 | 83 | 16600 |
41.2 | 1.02 |
Absolute molecular weights (MW) were determined in chloroform (a good solvent for both the lipid and the polymer) by size exclusion chromatography with multi-angle light scattering detection (SEC/MALLS). On the SEC chromatograms, the polymer peak exhibited an apparent trail on the low molecular weight side (Fig. S3†). However, this phenomenon was less important in chloroform compared to what was previously observed in THF22 and did not influence the MW determination thanks to the use of the MALLS detection. Since the MW values remained almost constant along that trail, it suggested the occurrence of some adsorption of the lipid–P(NAM-stat-NAS) chains onto the stationary phase of the column due to the lipid chain-end. As shown in Table 1, various LEP samples were synthesized from both A and B lipid-CTAs exhibiting MW between 5900 and 33000 g mol−1 with low dispersity (Đ) values.
We previously showed using MALDI-ToF mass spectrometry that a large majority of the polymer chains retained an intact lipid (coming from the lipid-CTA) at their α-end.22 This was further confirmed here by 1H NMR on a low MW conjugate (B1, 5900 g mol−1) (Fig. 3). Protons corresponding to the lipid-α-chain-end were clearly observed on top of the polymer signals, especially the protons of the fatty acid chains below 1.2 ppm. The more mobile methyl protons at the end of the fatty chains gave a more resolved peak compared to the broad peaks corresponding to the methylene protons. Integrals of these peaks were in agreement with the expected structure. In addition, the characteristic protons of the dithiobenzoate ω-chain-end were observed at 7.4, 7.6 and 8.0 ppm.
 | ||
| Fig. 3 1H NMR (500 MHz) spectrum of B1 LEP in CDCl3. | ||
Therefore, the lipid-copolymers exhibited the following features: (i) a controlled and adjustable MW, (ii) a narrow MW distribution (iii) a lipid at their α-end, (iv) multiple activated ester functions regularly spaced along the backbone and available for (bio)conjugation.
Synthesis and characterization of well-defined fluorescent LEPs
Fluorescent LEP conjugates were subsequently elaborated from these multifunctional LEPs using a two-step procedure. The first step consisted in the covalent coupling of a precise number of amino-functionalized chromophores onto the activated ester functions of the NAS units (Fig. 4). Thanks to the well-defined composition of the copolymer chains and to the efficiency of the coupling reaction (60–70% yields were generally obtained) (Table 2), the number of chromophores per polymer chain could be controlled by adjusting the stochiometry. Then, the second step consisted in a post-treatment of the unreacted activated ester functions along the copolymer chain. On a general point of view, this post-treatment step can be advantageously used to introduce other entities of interest along the backbone but also electrostatic charges. Here, two different post-treatments were performed, either a capping with aminoethylmorpholine (AEM) or a hydrolysis (leading to carboxylate charges) in order to increase the water-solubility and to obtain neutral and negatively charged conjugates, respectively (Fig. 4). | ||
| Fig. 4 Covalent coupling of the amino-derivatized chromophore 1 onto the activated lateral functions of the LEPs and post-treatment of the residual reactive functions of the copolymer by either basic hydrolysis or aminoethyl morpholine (AEM) capping. | ||
| Samplea | Mnb (g mol−1) | Coupling yieldc (%) | ncd | nCOO−e | dcf (%) |
|---|---|---|---|---|---|
| a H and AEM suffixes refer to hydrolyzed and AEM-capped conjugates, respectively.b Number average molecular weight of the fluorescent conjugates after post-treatment calculated assuming that the conjugates were in their sodium carboxylate form after dialysis and lyophilization.c Coupling yields determined by SEC/UV.39d nc is the average number of chromophores per polymer chain.e nCOO− is the maximum average number of carboxylate charges per polymer chain. The number of deprotonated COOH groups is variable, depending on the pH and on the distribution of these groups along the polymer backbone.f dc is the average density of chromophores per polymer chain. | |||||
| A1-2H | 6800 | 70 | 2.0 | 16.1 | 4.4 |
| A1-4H | 7500 | 72 | 4.1 | 14.0 | 9.1 |
| A2-4H | 16800 |
60 | 3.8 | 46.7 | 3.0 |
| A2-11H | 19200 |
70 | 10.8 | 39.8 | 8.5 |
| A3-1H | 25500 |
46 | 0.7 | 83.8 | 0.3 |
| A3-9H | 28300 |
65 | 8.5 | 76.0 | 4.0 |
| A3-9AEM | 36400 |
65 | 8.9 | 0 | 4.2 |
| A3-38H | 38000 |
63 | 37.8 | 46.7 | 17.9 |
| A3-38AEM | 43100 |
63 | 37.8 | 0 | 17.9 |
| B2-4H | 14000 |
68 | 3.9 | 37.3 | 3.8 |
A library of fluorescent LEP conjugates was obtained by varying the nature of the lipid at the α-chain-end (from A or B lipid-CTAs), the MW of the chains, the average number of chromophores per polymer chain (nc), and the type of post-treatment of the residual activated ester functions (Table 2). The coupling reaction was evidenced by 1H NMR (Fig. S4†) and SEC/UV that was used to monitor the coupling yield and thus nc (see Experimental section).39 Conjugate final MW ranged from 6800 to 43100 g mol−1 whereas nc was varied from 0.7 to 37.8. The average density of chromophores per chain, dc = 100 × nc/DPn (where DPn is the average degree of polymerization) was therefore between 0.3 and 17.9%. Finally, the average number of electrostatic charges per chain ranged from 0 for AEM-capped conjugates (except the phosphate charge of the phospholipid) up to 84 carboxylate groups for A3-1H hydrolyzed conjugate.
For this study, as explained in the Introduction, we used chromophore 1 (Fig. 4) that exhibits the distinctive property to be fluorescent in the crystalline state.35 Moreover, thanks to several other spectroscopic properties, this fluorophore is particularly well-suited for optical microscopy since it is characterized by (i) a large Stokes shift that helps to exclude the scattered and reflected light and to filter background fluorescence;41–43 (ii) a far-red fluorescence emission that fits with the common Cy5 emission filters. However, the free chromophore is hydrophobic and not water-soluble. One of the objectives here was to obtain water-soluble lipid–polymer probes by coupling chromophores 1 onto the lipid–P(NAM-stat-NAS) copolymers. This important feature would indeed be an opportunity to enlarge the use of this fluorophore for bioimaging applications.
In fact, the solubility of the fluorescent LEP conjugates depended on dc and on the type of post-treatment (capping or hydrolysis). Although the free chromophore was water-insoluble, LEP conjugates were soluble in aqueous media, except the conjugate A3-38AEM with a very high density of chromophores per chain (dc = 17.9%). Interestingly, the AEM-capped conjugates were soluble both in water and in chloroform, whereas the hydrolyzed conjugates were soluble in water and in polar organic solvents (such as ethanol and DMF).
Photophysical characterization of the fluorescent LEPs
The direct comparison of the spectroscopic properties of the hydrophobic free chromophore with the corresponding AEM-capped conjugates was possible in the same organic solvent (chloroform). Concerning the hydrolyzed conjugates, their properties were investigated in water (Table 3).| Sample | Solvent | Abs. λmax (nm) | Em. λmax (nm) | ε (M−1 cm−1) | ϕb | Brightness ε × ϕ |
|---|---|---|---|---|---|---|
| a Conjugate poorly soluble in water. It was thus not possible to determine the corresponding molar extinction coefficient.b Measured at λex = 510 nm using erythrosin B in MeOH (ϕr = 0.09) as the reference. | ||||||
| 1 | CHCl3 | 505 | 640 | 19000 |
0.07 | 1300 |
| A1-2H | Water | 506 | 688 | 32000 |
0.06 | 1900 |
| A1-4H | Water | 502 | 688 | 58000 |
0.03 | 1700 |
| A2-4H | Water | 508 | 688 | 56000 |
0.08 | 4500 |
| A2-11H | Water | 501 | 691 | 116000 |
0.03 | 3500 |
| A3-1H | Water | 509 | 688 | 12000 |
0.10 | 1200 |
| A3-9H | Water | 501 | 690 | 187000 |
0.07 | 13100 |
| A3-9AEM | CHCl3 | 505 | 645 | 214000 |
0.07 | 15000 |
| Water | 501 | 690 | 231000 |
0.02 | 4600 | |
| A3-38Ha | Water | 503 | 692 | n/a | 0.003 | n/a |
| A3-38AEM | CHCl3 | 502 | 650 | 839000 |
0.06 | 50300 |
| B2-4H | Water | 505 | 689 | 65000 |
0.05 | 3100 |
The maximum absorption wavelength of the bound chromophore (≈505 nm) remained almost unchanged compared to the free chromophore and was affected neither by the structure of the conjugates nor by the solvent (Fig. S5†). Absorption band was slightly broader in water than in chloroform (with a concomitant slight decrease of the molar extinction coefficient per bound chromophore). In contrast, the fluorescence emission spectrum was clearly red-shifted (by about 50 nm) in water compared to chloroform (due to the increased solvent polarity) (Fig. 5), whatever the structure and post-treatment. The large Stokes shift determined for the conjugates in chloroform (4430 ± 80 cm−1) was thus even larger in water (5700 ± 200 cm−1). As expected, fluorescence quantum yield (ϕ) of the bound fluorophore was found to decrease with dc (Fig. 6) due to fluorescence self-quenching, a well-known phenomenon associated with a high local chromophore concentration.44 In water, ϕ values were lower for AEM-capped conjugates compared to hydrolyzed conjugates (ϕ = 0.02 for A3-9AEM compared to 0.07 for A3-9H) probably reflecting that the hydrolyzed conjugates adopt a more expanded conformation (due to electrostatic repulsions between the carboxylate charges) that disfavors non-fluorescent dimer formation. Nevertheless, AEM-capped conjugates in chloroform and hydrolyzed conjugates (with dc < 4.5%) in water exhibited ϕ values that were very similar to the one of the free chromophore in chloroform. It has to be mentioned that it is quite exceptional that polymer–chromophore conjugates (especially in water) retain the same fluorescence quantum yield than that of the corresponding free chromophore in organic solvent. Here, it may be due to the unique properties of this chromophore derived from isophorone. Sample A3-1H, with on average less than 1 chromophore per chain, was expected to reflect the influence of the binding on the fluorophore properties (de-correlated from the mutual influence of neighboring fluorophores). Its ϕ value of 0.10 was the highest one, indicating a positive influence of the binding onto the polymer chain.
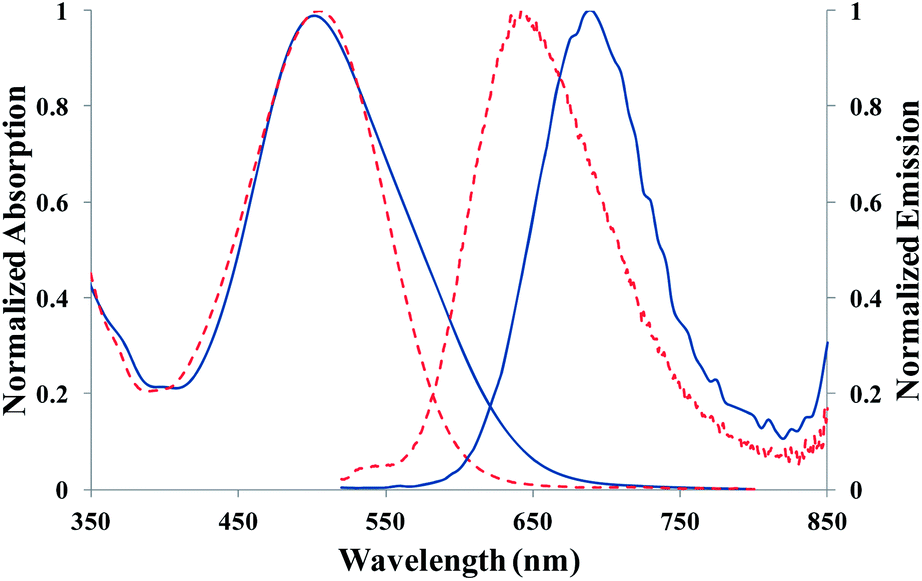 | ||
| Fig. 5 Absorption and emission spectra of A3-9AEM LEP in water (full lines) and in chloroform (dashed lines). | ||
 | ||
| Fig. 6 Fluorescence quantum yield in water versus the average fluorophore density along the polymer chain for the hydrolyzed conjugates. The fluorescence quantum yield of the free chromophore 1 in chloroform is represented as a dashed horizontal line. | ||
Consequently, besides providing water-solubility, multifunctional hydrophilic polymer chains led to far-red emitting conjugates with much improved brightness (B = εϕ) (Table 3). For instance, A3-38AEM conjugate was 38 fold brighter in chloroform than the free fluorophore and A3-9H conjugate was one order of magnitude brighter in water than the free fluorophore (in chloroform) (Fig. S6†).
Labeling of model lipid bi-layers by the fluorescent LEPs
In our previous study, it was shown that lipid-ended P(NAM) homopolymers were able to stabilize LipoParticle assemblies.22 Here, we used liposomes as model systems in order to test the ability of the fluorescent LEPs to be inserted into lipid bilayers (Fig. 7). | ||
| Fig. 7 Schematic representation of liposomes bearing chain-end anchored fluorescent LEPs. | ||
Two different approaches were explored by introducing the fluorescent LEP either before or after liposome formation. In the first approach, liposomes of various sizes were prepared from mixtures of natural lipids containing 0.1 and 1 mol% of fluorescent LEPs (e.g. A3-9H) using different conventional techniques (Experimental Part). Their sizes were assessed by dynamic light scattering (DLS).
First, small unilamellar vesicles (SUVs) were prepared by sonication or extrusion from various lipid mixtures (Table 4). They exhibited a narrow size distribution with diameters as low as 40 nm. The latter was smaller for sonicated vesicles than for extruded ones,45 and varied with lipid composition. Second, large unilamellar vesicles (LUVs), extruded at 50 °C through polycarbonate membranes with controlled porosities (from 50, 100, 200 nm up to 1 μm) gave reproducible results and narrow size distributions (Fig. S7†). Nevertheless, their measured diameters (e.g. 120 nm for LUV samples extruded through a 100 nm pore-size membrane) were slightly higher than the actual pore size of the membranes. Finally, giant unilamellar vesicles (GUVs) were obtained by electroformation38 from dioleyl phosphatidylcholine (DOPC) mixtures containing 0.1 mol% of A3-9H fluorescent LEP conjugate.46 They could be observed by optical microscopy since their diameter (>1 μm) was higher than the inherent resolution limit of conventional optical microscopes (>200–400 nm). Using dark-field microscopy, GUVs were visualized as white circles (freely moving in solution) with a 5–10 μm diameter. Fluorescence imaging performed on the same sample confirmed that the GUVs were highly fluorescent in the far-red range in the presence of the LEP conjugate whereas they remained dark in their absence (results not shown). Although the molar concentration of the conjugate into the lipid mixture was very low, the fluorescent GUVs were observed with a high signal-to-noise ratio thanks to the enhanced brightness of the probe.
| Composition (mol%) | Preparation | DLS diameter (nm) |
|---|---|---|
| a A3-9H conjugate.b C8-ceramide.c 50 nm pore-size membrane. | ||
| EggPC/PS/LEPa 80/20/0.1 | Sonication | 43 |
| DOPC/C8b/LEPa 80/20/0.1 | Sonication | 61 |
| DOPC/DOPE/LEPa 75/25/0.1 | Sonication | 74 |
| DOPC/Chol./LEPa 75/25/0.1 | Sonication | 61 |
| EggPC/PS/LEPa 80/20/0.1 | Extrusionc | 95 |
The second approach was designed to evaluate if the fluorescent LEPs were able to insert into pre-formed liposomes. For this two-step approach, DOPC GUVs were first prepared without conjugate and the absence of fluorescence was confirmed by fluorescence microscopy (Fig. 8, top panels). Then, after incubation of the pre-formed GUVs with an aqueous solution of A1-2H LEP conjugate (0.5 mol% compared to DOPC),47 the GUVs appeared strongly fluorescent, even more than the ones obtained via the first approach (Fig. 8, low panels).
 | ||
| Fig. 8 Dark field (left, A and C) and fluorescence (right, B and D) microscopy images of GUVs obtained by the electroformation process, before (top images, A and B) and after incubation with A1-2H LEP conjugate (bottom images, C and D). | ||
Those results confirmed that the fluorescent LEPs were able to insert into lipid bilayers. It is however noteworthy that the intrinsic spatial resolution of fluorescence microscopy is by far not sufficient to firmly affirm that they were anchored in an oriented manner through their lipid α-end. Nevertheless, this assay suggests that the new fluorescent lipid–polymer probes are powerful tools to label the surface of lipid self-assemblies.
Conclusions
The multifunctional lipid-ended polymers, LEPs, reported here constitute a new class of lipid–polymer conjugates (in comparison with lipid-homopolymers like lipid–PEGs), displaying multiple reactive sites regularly distributed along the polymer chains. Their elaboration was based on the synthesis of lipid-functionalized RAFT agents that were successfully used to control the copolymerization of NAM and NAS monomers at the azeotropical composition. The activated ester functions of the resulting lipid–P(NAM-stat-NAS) conjugates enable an efficient coupling of a variety of amino-derivatized (bio)-compounds along the polymer backbone.Here, we developed highly fluorescent LEPs by coupling multiple copies of a far-red emitting chromophore (from 1 to 38) on different lipid–P(NAM-stat-NAS) backbones. Although the free fluorophore was not water-soluble, the resulting conjugates were soluble in aqueous media. Their thorough spectroscopic characterization showed that coupling multiple fluorophores along the polymer chain resulted in probes with an enhanced brightness, up to 10 fold in water.
As a first proof of concept, we evidenced that these new fluorescent lipid probes can be inserted into pre-formed lipid bilayers of liposomes of various sizes. Thus, they are very promising tools for the fluorescent labeling of different kinds of lipid self-assemblies. The study of their behavior in cellulo will be reported shortly.
More generally, the well-controlled and modular architecture of the multifunctional LEPs that were designed, paved the way towards the elaboration of a large range of functionalized lipid–polymer conjugates. The nature of the lipid, the chain length, the nature of the ω-end group as well as the number and the nature of the bound molecules can be tuned. This modularity thus offers countless opportunities to functionalize the surface of biomimetic lipid membranes, in applications such as liposome-mediated drug delivery.
Experimental section
Materials
All chemicals were purchased from Sigma-Aldrich, Acros and Fluka at the highest purity available and used without further purification. THF and 1,4-dioxane were distilled over LiAlH4. N-Acryloyl morpholine (NAM) (Aldrich, 97%) was distilled under reduced pressure (120 °C; 10 mmHg) to remove inhibitor. N-Acryloxysuccinimide (NAS) was synthesized as previously described.27 2,2′-Azobis(isobutyronitrile) (AIBN) was purified by recrystallization from ethanol. Other solvents were used as received from the supplier without further purification. All solvents used for determination of the photophysical properties were of spectrophotometric grade. Lipids including dipalmitoylphosphoethanolamine (DPPE), phosphatidylcholine (PC), phosphatidylserine (PS), dioleolylphosphocholine (DOPC), C8-ceramide and cholesterol (Chol) were purchased from Avanti Polar Lipids and Sigma-Aldrich.Synthesis and purification of lipid-CTAs
Lipid-functionalized chain transfer agents (lipid-CTAs) were synthesized from succinimido-2-[[2-phenyl-1-thioxo]thio]-propanoate (SEDB)36 following the procedure described by Bathfield et al.22 Purification protocols for both SEDB and the lipid-CTAs were however improved compared to the original procedure as explained below.After SEDB synthesis from CAEDB, the reaction mixture was filtrated. After solvent removal, the product was twice dissolved in a minimum volume of ethyl acetate and filtrated. The product was finally purified by re-cristallization in a mixture of chloroform–pentane (10/90 vol%) leading to a pink solid with a >95% purity (1H NMR) (final yield after purification: 35%).
The newly synthesized B lipid-CTA was obtained using this protocol with DPPE as the lipid:
1H NMR 200 MHz (CDCl3, 300 K): δ (ppm): 0.88 (6H); 1.24 (48H); 1.56 (4H); 1.63 (3H); 2.26 (4H); 3.47 (2H); 3.91 (4H); 4.10 (1H); 4.34 (1H); 4.69 (1H); 5.18 (1H); 7.35 (dd, 2H); 7.51 (dd, 2H); 7.97 (d, 2H).
ESI-ToF mass spectrometry (microToF QII Bruker Daltonics): characteristic ion [M − H]−, C47H81NO9PS2; calculated 898.5090 mass units; found 898.5073 mass units.
Synthesis of multifunctional LEP conjugates
Poly(NAM-stat-NAS) copolymer synthesis in the presence of the lipid-CTA was adapted from the procedure previously described for the homopolymerization of NAM.22 The initial co-monomer molar ratio was the azeotropical composition 60/40 mol% NAM/NAS.Briefly, NAM (1.112 g, 7.88 mmol), NAS (0.888 g, 5.25 mmol), lipid-CTA A (103.2 mg, 0.115 mmol), AIBN (3.44 mg, 0.021 mmol), dioxane (5.57 mL), and trioxane (0.056 g, internal reference for 1H NMR determination of monomer consumption) were introduced in a Schlenk tube equipped with a magnetic stirrer. The mixture was degassed by 3 freeze–evacuate–thaw cycles and then heated under nitrogen using a thermostated oil bath (80 °C). Periodically, samples were withdrawn from the polymerization medium for analyses. Polymers were precipitated in a large volume of diethyl ether, recovered by centrifugation, and finally dried under vacuum.
Synthesis of fluorescent LEP conjugates
25 mg of lipid–P(NAM-stat-NAS) copolymer were dissolved in 1 mL of chloroform in a 25 mL round bottom flask equipped with a magnetic stirrer. Then, 3.6 mg of 1 dissolved in chloroform (9.7 × 10−2 M) were added together with 2 equivalents of triethylamine (Et3N). Polymer concentration was adjusted to 10 mg mL−1 with chloroform and the coupling reaction was carried out at 40 °C in the dark under stirring for 24 hours. The coupling yield was followed by SEC/UV measurements and was typically between 60 and 70%. The red LEP conjugate was then precipitated in a large volume of diethyl ether and isolated by centrifugation. The procedure was repeated until complete discoloration of the supernatant, indicating the removal of the free unreacted fluorophore. Purified conjugates were finally dried under vacuum up to constant weight.
AEM capping. 25 mg of conjugates were re-dispersed in 3 mL of chloroform before addition of a 10-fold molar excess of AEM (compared to the initial NAS units). The capping reaction proceeded at room temperature under magnetic stirring overnight. The conjugates were then isolated by precipitation in diethyl ether before dialysis against deionized water (Spectrum Labs, Spectra/Por 6, MWCO: 2 kg mol−1) and finally dried by lyophilization.
Hydrolysis. 45 mL of a borate buffer (50 mM, pH = 9) were added to 25 mg of conjugates. Hydrolysis proceeded at room temperature under magnetic stirring for 3 days. After hydrolysis, conjugates were purified by dialysis against deionized water (Spectrum Labs, Spectra/Por 6, MWCO: 2 kg mol−1) and dried by lyophilization.
Liposome model systems
Large and small unilamellar vesicles, LUVs and SUVs respectively, were prepared by extrusion.37 Mixtures in water of EggPC (160 μL of a 10 mg mL−1 solution), BrainPS (40 μL of a 10 mg mL−1 solution) (80/20 mass%) and the fluorescent LEP conjugate (0.1 mol% of the total lipids) were dried in a Speedvac rotary evaporator overnight. The dry lipid film was then hydrated with a PBS buffer pH 7.4 for 2 hours at 45 °C, with vortexing every 15 minutes. 12 freeze–thaw cycles were performed by freezing the vesicle solutions in liquid nitrogen followed by thawing (water bath, 40 °C). These solutions were finally extruded at 50 °C, through controlled pore-size polycarbonate membranes (Whatman Nucleopore Track-Etch, 19 mm), using an Avanti Polar Lipids miniextruder.Small unilamellar vesicles, SUVs, were also prepared by sonication of lipid mixtures in PBS solution (10 mg mL−1) using a Bioruptor®Plus (UCD-300) from Diagenode. Solutions were maintained at 60 °C before sonication (12 × 6 cycles of 1 minute – 320 W).
Giant unilamellar vesicles, GUVs, were prepared by electroformation following the procedure described by Portet et al.38 from a 0.5 mg mL−1 chloroform–ethanol 90/10 vol% solution of DOPC containing 0.1 mol% (compared to DOPC) of fluorescent LEP conjugate. To test the ability of the fluorescent LEP conjugates to insert into pre-formed GUVs, 100 mol% DOPC GUVs were first prepared using the above-mentioned procedure, before incubation with an aqueous solution of LEP conjugate (A1-2H, 0.5 mol% compared to DOPC) under gentle stirring for 2 hours at 37 °C.
Characterization techniques
Acknowledgements
We acknowledge the contribution of Jean-Michel Lucas and Agnès Crépet (Laboratoire d'Ingénierie des Matériaux Polymères) for technical support in SEC/MALLS measurements, Sandrine Denis-Quanquin (Laboratoire de Chimie, ENS de Lyon) for NMR measurements and Elise Hamard-Péron for her help with the liposome extrusion technique. SA acknowledges a PhD grant from the Région Rhône-Alpes, Cluster 5 Chimie. WL was supported by an AMI grant from the French Bioimaging network, GDR 2588 MiFoVI-CNRS. We also thank the Fondation Innovations en Infectiologie, FINOVI, and the CNRS interdisciplinary program (PIR051) for funding.Notes and references
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145 CrossRef CAS PubMed.
- J. C. Su, C. L. Tseng, T. G. Chang, W. J. Yu and S. K. Wu, Bioorg. Med. Chem. Lett., 2008, 18, 4593 CrossRef CAS PubMed.
- S. Zalipsky, B. Puntambekar, P. Boulikas, C. M. Engbers and M. C. Woodle, Bioconjugate Chem., 1995, 6, 705 CrossRef CAS PubMed.
- G. Bendas, A. Krause, U. Bakowsky, J. Vogel and U. Rothe, Int. J. Pharm., 1999, 181, 79 CrossRef CAS PubMed.
- K. Edwards, M. Johnsson, G. Karlsson and M. Silvander, Biophys. J., 1997, 73, 258 CrossRef CAS PubMed.
- N. Dos Santos, C. Allen, A. M. Doppen, M. Anantha, K. A. K. Cox, R. C. Gallagher, G. Karlsson, K. Edwards, G. Kenner, L. Samuels, M. S. Webb and M. B. Bally, Biochim. Biophys. Acta, 2007, 1768, 1367 CrossRef CAS PubMed.
- S. Sriwongsitanont and M. Ueno, Chem. Pharm. Bull., 2002, 50, 1238 CrossRef CAS PubMed.
- A. L. Klibanov, K. Maruyama, V. P. Torchilin and L. Huang, FEBS Lett., 1990, 268, 235 CrossRef CAS PubMed.
- S. Unezaki, K. Maruyama, O. Ishida and A. Suginaka, Int. J. Pharm., 1995, 126, 41 CrossRef CAS.
- V. P. Torchilin, T. S. Levchenko, A. N. Lukyanov, B. A. Khaw, A. L. Klibanov, R. Rammohan, G. P. Samokhin and K. R. Whiteman, Biochim. Biophys. Acta, 2001, 1511, 397 CrossRef CAS.
- S. Zalipsky, Nat. Rev. Drug Discov., 1995, 16, 157 CAS.
- M. T. Allen, E. Brandeis, C. B. Hansen, G. Y. Kao and S. Zalipsky, Biochim. Biophys. Acta, 1995, 1237, 99 CrossRef.
- F. Albertorio, A. J. Diaz, T. Yang, V. A. Chapa, S. Kataoka, E. T. Castellana and P. S. Cremer, Langmuir, 2005, 21, 7476 CrossRef CAS PubMed.
- G. A. F. Van Tilborg, E. Vucic, G. J. Strijkers, D. P. Cormode, V. Mani, T. Skajaa, C. P. M. Reutlingsperger, Z. A. Fayad, W. J. M. Mulder and K. Nicolay, Bioconjugate Chem., 2010, 21, 1794 CrossRef CAS PubMed.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458 CrossRef CAS.
- A. Favier and M. T. Charreyre, Macromol. Rapid Commun., 2006, 27, 653 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS PubMed.
- K. Ohno, T. Fukuda and H. Kitano, Macromol. Chem. Phys., 1998, 199, 2193 CrossRef CAS.
- H. Götz, E. Harth, S. M. Schiller, C. W. Frank, W. Knoll and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3379 CrossRef.
- P. Kujawa, F. Segui, S. Shaban, C. Diab, Y. Okada, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 341 CrossRef CAS.
- M. Bathfield, D. Daviot, F. D'Agosto, R. Spitz, C. Ladavière, M. T. Charreyre and T. Delair, Macromolecules, 2008, 41, 8346 CrossRef CAS.
- L. Hespel, E. Kaifas, L. Lecamp, L. Picton, G. Morandi and F. Burel, Polymer, 2012, 53, 4344 CrossRef CAS.
- A. Favier, B. De Lambert and M. T. Charreyre, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, 2008, p. 556 Search PubMed.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS.
- J. Hwang, R. C. Li and H. D. Maynard, J. Controlled Release, 2007, 122, 279 CrossRef CAS PubMed.
- A. Favier, F. D'Agosto, M. T. Charreyre and C. Pichot, Polymer, 2004, 45, 7821 CrossRef CAS.
- O. Maier, V. Oberle and D. Hoekstra, Chem. Phys. Lipids, 2002, 116, 3 CrossRef CAS PubMed.
- J. A. Monti, S. T. Christian and W. A. Shaw, J. Lipid Res., 1978, 19, 222 CAS.
- A. Chaudhary, J. Chen, Q. M. Gu, W. Witke, D. J. Kwiatkowski and G. D. Prestwich, Chem. Biol., 1998, 5, 273 CrossRef CAS PubMed.
- R. Koynova, H. S. Rosenzweig, L. Wang, M. Wasielewski and R. C. MacDonald, Chem. Phys. Lipids, 2004, 129, 183 CrossRef CAS PubMed.
- W. Qin, D. Ding, J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 771 CrossRef CAS.
- V. Pansare, S. Hejazi, W. Faenza and R. K. Prud'homme, Chem. Mater., 2012, 24, 812 CrossRef CAS PubMed.
- J. Massin, A. Charaf-Eddin, F. Appaix, Y. Bretonnière, D. Jacquemin, B. Van der Sanden, C. Monnereau and C. Andraud, Chem. Sci., 2013, 4, 2833 RSC.
- J. Massin, W. Dayoub, C. Mulatier, C. Aronica, Y. Bretonnière and C. Andraud, Chem. Mater., 2011, 23, 862 CrossRef CAS.
- M. Bathfield, F. D'Agosto, R. Spitz, M. T. Charreyre and T. Delair, J. Am. Chem. Soc., 2006, 128, 2546 CrossRef CAS PubMed.
- G. Blin, E. Margeat, K. Carvalho, C. A. Royer, C. Roy and C. Picart, Biophys. J., 2008, 94, 1021 CrossRef CAS PubMed.
- T. Portet, F. C. i. Febrer, J. M. Escoffre, C. Favard, M. P. Rols and D. S. Dean, Biophys. J., 2009, 96, 4109 CrossRef CAS PubMed.
- C. Cepraga, T. Gallavardin, S. Marotte, P. H. Lanoë, J. C. Mulatier, F. Lerouge, S. Parola, M. Lindgren, P. L. Baldeck, J. Marvel, O. Maury, C. Monnereau, A. Favier, C. Andraud, Y. Leverrier and M. T. Charreyre, Polym. Chem., 2013, 4, 61 RSC.
- N. Boens, W. Qin, N. Barasić, J. Hofkens, M. Ameloot, J. Pouget, J. P. Lefèvre, B. Valeur, E. Gratton, M. VandeVen, N. D. Silva Jr, Y. Engelborghs, K. Willaert, A. Sillen, A. J. W. G. Visser, A. Van Hoek, J. R. Lakowicz, H. Malak, I. Gryczynski, A. G. Szabo, D. T. Krajcarski, N. Tami and A. Miura, Anal. Chem., 2007, 79, 2137 CrossRef CAS PubMed.
- A. J. Amoroso, M. P. Coogan, J. E. Dunne, V. Fernández-Moeira, J. B. Hess, A. J. Hayes, D. Lloyd, C. Millet, S. J. A. Pope and C. Williams, Chem. Commun., 2007, 3066 RSC.
- A. N. Glazer, J. Appl. Phycol., 1994, 6, 105 CrossRef CAS.
- K. D. Piatkevich, J. Hulit, O. M. Subach, A. Abdulla, J. E. Segall and V. V. Verkhusha, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5369 CrossRef CAS PubMed.
- J. Baumann and M. D. Fayer, J. Chem. Phys., 1986, 85, 4087 CrossRef CAS.
- L. T. Boni, M. M. Batenjany, M. E. Neville, Y. Guo, L. Xu, F. Wu, J. T. Mason, R. J. Robb and M. C. Popescu, Biochim. Biophys. Acta, 2001, 1514, 127 CrossRef CAS.
- Y. Yamashita, M. Oka, T. Tanaka and M. Yamazaki, Biochim. Biophys. Acta, 2002, 1561, 129 CrossRef CAS.
- D. Cuvelier, C. Vezy, A. Viallat, P. Bassereau and P. Nassoy, J. Phys.: Condens. Matter, 2004, 16, S2427 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Complementary physico-chemical and photo-physical characterizations of lipid-CTAs (1H NMR), multifunctional LEPs (SEC chromatogram, 1H NMR), fluorescent LEPs (absorption and emission spectra of A3-9AEM and chromophore 1 in chloroform, brightness of chloroform-soluble compounds) and liposomes (size distribution measured by DLS for A3-9H). See DOI: 10.1039/c4ra01334d |
| ‡ Present address: Centre d'études d'agents Pathogènes et Biotechnologies pour la Santé, CNRS UMR 5236, F-34293, Montpellier, France. |
| This journal is © The Royal Society of Chemistry 2014 |