
Physicochemical investigations of the metal complexes of l-valine with doubly charged ions of nickel, copper and zinc: a combined experimental and computational approach†
Abstract
Assessment of the intrinsic structural changes inflicted by metal coordination in the molecular geometries of the genetically encoded amino acids is of fundamental importance for proper characterization and understanding of the structure–function relationships of proteins and polypeptides. Efforts are being made here to investigate the coordination properties of L-valine as a potential metal-binding entity involving a combined solid state solvent free synthetic and computational approach. The metal complexes of L-valine with the divalent cations of nickel, copper and zinc are characterized by elemental analyses, molar conductance, EDAX-SEM, TEM, TG/DTA, infrared, electronic absorption, fluorescence and mass spectroscopy. Quantum chemical calculations carried out in gas and aqueous phase using the B3LYP/6-311++G(d,p) level of theory provide valuable insights concerning the interaction enthalpies and Gibbs energies; vibrational and absorption spectra along with various other molecular and electronic properties of the metal complexes. Solvation effects are evident on the structure and stability of the metal complexes. Analysis of the geometrical parameters suggests significant changes in the molecular structure of L-valine as a result of metal binding. The harmonic frequencies furnished at the B3LYP level are in good agreement with the earlier theoretical and experimental observations and well reflect the influence of the aqueous environment and identity of the metal ions on the structural and molecular properties of the complexes. The physical origin of the molecular interactions of L-valine with the metal ions has also been evaluated by performing EDA.
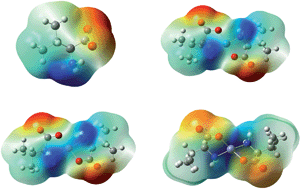
 Please wait while we load your content...
Please wait while we load your content...

