DOI:
10.1039/C4RA01222D
(Paper)
RSC Adv., 2014,
4, 27843-27849
Integrated lignin-mediated adsorption-release process and electrochemical reduction for the removal of trace Cr(VI)
Received
11th February 2014
, Accepted 11th June 2014
First published on 11th June 2014
Abstract
Hexavalent chromium Cr(VI) is extremely toxic and is classified as a human carcinogen, even at trace concentrations. Hence, it is critical to develop reliable and efficacious methods for its elimination. Here we report on the development of an effective lignin-mediated adsorption-release process based upon the pH-dependent solubility of lignin to significantly enhance the electrochemical removal of trace Cr(VI). The adsorption and pre-concentration of Cr(VI) was carried out at pH 2.0, while the release and electrochemical reduction was performed at pH 11.0. The 100% efficient release process was due to the complete dissolution of lignin within alkaline solution. The released Cr(VI) ions were electrochemically reduced, forming insoluble Cr(OH)3, which could be easily separated. This novel approach overcomes the drawback of non-degradability in adsorption by the latter electrochemical reduction phase and the efficiency limitation of electrochemical process by the prior preconcentration step. The new integrated lignin-mediated adsorption-release and electrochemical reduction process proposed in this study comprises a promising strategy for the removal of trace concentrations of Cr(VI).
1. Introduction
Recently, considerable attention has been focused on heavy metal (Cr, Cu, Hg, Pb and As) pollution abatement, due to severe toxicity within ecosystems and to human health.1 Among these heavy metal effluents, chromium species, which are discharged from various industrial processes, stand out as some of the most prominent hazardous contaminants.2 There are two stable Cr oxidation states, Cr(III) and Cr(VI), which present substantially different toxicity and physicochemical properties.3 Cr(III) compounds are considered to be harmless with limited mobility;4 whereas Cr(VI) compounds are extremely toxic and classified as a human carcinogen, even at trace concentrations.5 Furthermore, Cr(VI) compounds are highly soluble in most aqueous and biochemical systems.6,7
Toward the reduction of Cr(VI) to recommended levels, several treatment techniques have been attempted. Wastewater containing Cr(VI) is conventionally treated via the chemical reduction of Cr(VI) to Cr(III) using SO2 gas or NaHSO3, followed by pH-dependent precipitation to insoluble Cr(III) hydroxide.8 The disadvantages of this process include the large volume of reducing agents consumed and residual sludge discharges.9 Moreover, this technique is suitable only for the treatment of wastewater that contains high Cr(VI) concentrations, and the residual Cr(VI) is generally >8.7 ppm due to its low reduction efficiency.10 Alternatively, electrochemical methods have gained increasing attention because of their operational versatility and “clean” properties.11,12 The removal of Cr(VI) might be facilitated by electrogenerated Fe2+ and Al3+ at sacrificial Fe, Al electrodes (electrocoagulation)13,14 or by electrochemical reduction in acidic and alkaline solutions (eqn (1) and (2)).15–17 However, those methods still suffer from low efficiencies, and require relatively high initial Cr(VI) concentrations.11
| | |
HCrO4− + 7H+ + 3e− → Cr3+ + 4H2O
| (1) |
| | |
CrO42− + 4H2O + 3e− → Cr(OH)3 + 5OH−
| (2) |
In the interim, a wide range of techniques for Cr(VI) removal have been explored, such as ion exchange,18,19 membrane separation,20 biodegradation21 and adsorption.22 Among these methods, it has been considered that adsorption is an efficient and cost-effective approach, even for the removal of trace amounts of Cr(VI).23,24 Hence, significant efforts have been made in the development of potential adsorbents for the removal of Cr(VI), including activated carbons,25 agricultural by-products26 and polymeric materials.27 Most recently, there has been an increasing interest in low cost lignin-based adsorbents.28 Lignin is generated at a rate of 50 million tons per year from wood residues (e.g., sawdust and paper mill discharges) and agricultural residues (e.g., wheat bran, corn stoves, etc.).29 This attention is garnered not only because lignin has a comparable adsorption capacity to other natural sorbents, but is also due to its great beneficial impact as a value-added biosorbent, rather than a discarded solid waste.30 Lignin is three dimensional, extensively branched polyphenolic plant derived polymer. Its relatively low solubility in acidic solution31 and the presence of a large population of various oxygen-containing groups within the surface structure, enable sorption performance via the mechanism of physical adsorption, hydrogen bonding, as well as coordination and covalent-linking.32–34 However, the adsorption process transfers only the Cr(VI) from an aqueous solution to a solid phase (lignin), while no degradation to Cr(III) is achieved, thus further development is necessitated.35
Here we report on an novel lignin-mediated adsorption-release process for effective electrochemical removal of trace Cr(VI). It is known that lignin solubility is greatly pH-dependent.36 It is insoluble in acidic solutions, which is favorable for the adsorption of Cr(VI). Conversely, it is highly soluble in alkaline solutions, which allows complete release of adsorbed Cr(VI) and the direct electrochemical reduction of Cr(VI) to Cr(III) precipitate, as depicted in eqn (2). In the present study, the effect of the pH on the adsorption of Cr(VI) onto lignin has been investigated. The release of the adsorbed Cr(VI) was carried out at pH 11.0 and Au nanoparticles (NPs) supported on TiO2 nanotube (NT) arrays were used as the electrode for the electrochemical reduction of Cr(VI). The novel approach proposed in this study overcomes the drawback of non-degradability in adsorption by subsequent electrochemical reduction and the efficiency limitation of the electrochemical process that is imposed by the preconcentration step.
2. Materials and methods
2.1 Materials
All of the chemical reagents employed in this study were of analytic grade purity and used as received from Sigma-Aldrich, including hydrofluoric acid (HF, 48 wt% in H2O), dimethyl sulfoxide (DMSO, ≥99.5 wt%), gold(III) chloride hydrate (99.8 wt%), methanol (99.8 wt%), sulfuric acid (95.0–98.0 wt%), sodium chromate (Cr(VI), 98.0 wt%) and sodium hydroxide (≥98 wt%). Ultrapure water (18.2 MΩ cm) was obtained from a NANO pure® Diamond™ UV water purification system. The kraft lignin used in this study was extracted from black liquor, provided by a local pulp and paper mill, using carbon dioxide (CO2). The precipitated lignin was subsequently purified by rinsing with a dilute sulphuric acid solution. The electrolytes used in this research were prepared via the dissolution of corresponding chemicals in ultrapure water.
2.2 Fabrication of the Ti/TiO2NT/Au electrode
The Ti/TiO2NT/Au electrodes were prepared by a two-step process involving anodic oxidation followed by photo-assisted deposition.37–39 A fresh Ti substrate (1 cm2) was cleaned via sonication in acetone for 10 min followed by 10 min of immersion in ultrapure water, after which it was chemically etched in an 18% HCl solution at 85 °C for 25 min. After being rinsed with ultrapure water and dried in an Ar stream, the etched Ti substrate was anodized in a 2% (wt) HF + DMSO solution with a counter electrode comprised of platinum foil. Anodization was performed at 40 V for 8 h with a DC power supply, after which the sample was annealed at 450 °C for 3 h to obtain an anatase structure. For the decoration of Au NPs, the TiO2 nanotube array was placed in a deaerated 5 mL 50% (v/v) methanol solution that contained 0.3 mM HAuCl4. Subsequently, photo-assisted deposition was carried out under UV light illumination for 15 min. using a BlueWave TM 50 AS UV spot lamp (365 nm emission) with an intensity of ca. 20 mW cm−2. Electrons and holes were generated under the UV irradiation; and the Au(III) ions were reduced by the photo-generated electrons, forming Au nanoparticles on the TiO2 nanotubes. Finally, the electrode was rinsed with ultrapure water and dried in a vacuum oven at 40 °C. The as-prepared electrode surface morphology and composition were characterized by a Hitachi SU-70 Field emission scanning electron microscope (FE-SEM) with X-ray energy dispersive spectrometry (EDS), while its crystalline structure was determined using an X'Pert PANanalytical Diffractometer with Cu Kα radiation.
2.3 Lignin-mediated adsorption and release of Cr(VI)
The adsorption performance of Cr(VI) was studied by immersing 60 mg of lignin in 200 mL of a Cr(VI) solution (2 ppm) under continuous stirring at 25 °C. After different contact times or experimental conditions, the suspension was centrifuged, whereafter the solution was analyzed using UV-Visible spectroscopy (Cary 50, Agilent Technologies). For comparison, the Cr(VI) concentration was also measured using inductively coupled plasma atomic emission spectroscopy (ICP-AES, Varian Vista Pro Radial). After the adsorption procedure, the solid phase was separated, followed by its introduction into a 20 mL alkaline solution (pH = 11.0). The as-prepared solution was thoroughly stirred to reach the dissolution equilibrium.
2.4 Electrochemical reduction of Cr(VI)
The electrochemical measurements as well as electrochemical reduction of Cr(VI) was performed using a CHI (660B) workstation. The as-prepared Ti/TiO2NT/Au electrode was utilized as the working electrode, while the counter and reference electrodes were comprised of a Pt mesh and an Ag/AgCl (KCl saturated) electrode, respectively. Prior to each measurement, the electrolytes were deaerated with Ar for 20 min and then maintained under an Ar atmosphere for the duration of the measurements at room temperature (25 °C). The Ti/TiO2NT/Au electrode was cycled in stationary solution with its corresponding scan rate and voltage range to obtain cyclic voltammetric curves, while amperometric measurements were carried out in a stirred solution at 300 rpm with a fixed potential. It should be noted that the Cr(VI) concentration in this section was determined using ICP-AES due to the interference effect of lignin toward UV-Visible spectra.40
3. Results and discussion
3.1 Characterization of fabricated Ti/TiO2NT/Au electrode
Our recent studies have revealed that there is a significant improvement in activity for the electrochemical reduction of Cr(VI) at a Ti/TiO2NT/Au electrode as compared with a polycrystalline gold electrode, as the latter's performance is contingent on its nanoparticle/nanotubular heterojunction architectures.17 As shown in Fig. 1, a well-organized array of nanotubes (NTs) with an open top infrastructure was grown directly onto a Ti substrate via anodic oxidation. The uniform diameter of these nanotubes was ∼200 nm, with a wall thickness of ∼80 nm and length of ∼4–5 μm estimated from the cross-section SEM image. The EDS of Fig. 2a exhibited strong Ti and O peaks, and the corresponding XRD peaks as shown in Fig. 2b were identified as tetragonal anatase, indicating that the as-prepared nanotubes were anatase TiO2. Additionally, the gold nanoparticles of ∼20 nm in diameter were well decorated on the formed TiO2 NTs, as illustrated in Fig. 1. This was further confirmed by their corresponding EDS and XRD peaks in Fig. 2a and b, respectively. It should be noted that a small volume of ∼40 nm diameter Au nanoparticles were dispersed on the relatively longer nanotubes due to the aggregating nature of the Au nanoparticles (NPs).41 The formed Au nanoparticles were very stable on the surface of the TiO2 nanotubes. It was clear that the as-prepared Au nanoparticle TiO2 nanotube arrays were highly ordered with metal–semiconductor heterojunction infrastructures and high surface areas, which suggested potentially excellent electrochemical performance.
 |
| | Fig. 1 SEM images of the as prepared Au nanoparticle modified TiO2 nanotube electrode. | |
 |
| | Fig. 2 (a) EDX and (b) XRD spectra of the as prepared Ti/TiO2NT/Au electrode. | |
3.2 Electrochemical reduction of trace Cr(VI)
The cyclic voltammetric (CV) measurements were carried out in an acidic solution to determine the electrochemical reduction performance of trace Cr(VI) at the Ti/TiO2NT/Au electrode. As shown in Fig. 3a, a relatively weak and broad reduction peak emerged at 0.18 V, with a current density of 44 μA cm−2 in the presence of 2 ppm Cr(VI). No reduction peak was observed in the same potential region of its corresponding blank CV curve without Cr(VI), which indicated that this peak was attributed to Cr(VI) reduction. Further, there was no corresponding oxidation peak in the reverse scan of the CV curve in the presence of Cr(VI), which suggested an electrochemically irreversible reaction. The slight downward shift of the CV curve as compared to its blank curve might be attributed to the effect of the Cr(VI) reduction wave.42 Clearly, although the Ti/TiO2NT/Au electrodes possess excellent electrochemical activity toward the reduction of Cr(VI),17 their practical application for the removal of Cr(VI) is still limited by the low concentration of the trace Cr(VI).
 |
| | Fig. 3 (a) Cyclic voltammograms recorded at Ti/TiO2NT/Au electrode in the absence and presence of 2.0 ppm Cr(VI), pH = 2.0; (b) removal of Cr(VI) by electrochemical (EC) treatment at Ti/TiO2NT/Au electrode, potential = 0.18 V, inset: the comparison of UV-Vis spectroscopy via before and after treatment, pH = 2.0. | |
To further characterize the electrochemical removal of trace Cr(VI), chronoamperometry was employed and the measurement was conducted at a constant potential of 0.18 V, which is the peak potential seen in the reduction of Cr(VI) in Fig. 3a. As shown in Fig. 3b, an initial current density of 55 μA cm−2 was observed in the stirring solution. Additionally, a gradual decay of cathodic current density with increasing reaction time was observed, which might be due to the decrease of the concentration of Cr(VI) and the potential poisoning effect on the electrode surface. It has been shown the reaction product of Cr3+, as mentioned in eqn (1), could be adsorbed on the gold electrode surface.42 Therefore, Cr3+ would tenaciously begin to accumulate when its removal reaction could not keep pace with the continuous reduction of Cr(VI), leading to the decay of the Cr(VI) reduction current density. Consequently, as shown in the UV-Vis spectra (inset of Fig. 3b), the removal efficiency of the electrochemical reduction of trace Cr(VI) was found to be only 22% subsequent to a two-hour treatment time.
3.3 Adsorption of trace Cr(VI) by lignin
3.3.1 Effect of the pH on Cr(VI) adsorption. The pH of aqueous media is of great importance for the Cr(VI) adsorption process, since the pH influences not only the Cr speciation, but also the surface properties of the adsorbent.30,33 As shown in Fig. 4, the role of pH on the adsorption of Cr(VI) onto lignin was determined by varying the initial pH of solution, which ranged from 1.0 to 7.0. The optimized removal performance was achieved at a pH of 2.0. This phenomenon was due to the competition effect between surface exchange reactions and Cr(VI) speciation. It has been demonstrated Cr(VI) adsorption onto lignin originated mainly from the electrostatic attraction between negatively charged Cr(VI) ions and protonated (positively charged) functional groups that were present on the adsorbent surface.30 The higher the proton concentration of the aqueous solution, the better the H+ association was toward carboxyl, phenolic and hydroxyl groups on the lignin surface,33 which led to the increasing efficiency of Cr(VI) adsorption. In addition, the decrease of adsorption capacity from pH 2.0 to 1.0 was possibly due to pH-dependent Cr(VI) speciation,43 where Cr(VI) was transferred from HCrO4− and Cr2O72− to neutral H2CrO4, thereby decreasing the affinity between Cr(VI) and the surface charged lignin adsorbent. Consequently, a pH of 2.0 was selected as the optimized value for the following investigations.
 |
| | Fig. 4 The effect of pH on the adsorption of 2.0 ppm Cr(VI) by 0.3 g L−1 lignin for a two-hour period. | |
3.3.2 Adsorption isotherm. In order to determine the adsorption properties of lignin toward Cr(VI), the adsorption isotherm at a pH of 2.0 was measured by altering the initial Cr(VI) concentration, from 1 to 20 ppm, and the data was fitted utilizing the common Langmuir isotherm model as follows:35| |
 | (3) |
where the Ce (mg L−1) is the equilibrium Cr(VI) concentration, qe (mg g−1) is the equilibrium adsorption capacity of lignin, qm (mg g−1) is the maximum adsorption capacity of lignin, and K is the Langmuir isotherm constant. As shown in Fig. 5, the experimental data was fitted excellently by the Langmuir model (R2 = 0.998), suggesting a monolayer adsorption process. Further, the maximum adsorption capacity qm was calculated to be 33.33 mg g−1, which was well comparable to the value previously reported (31.6 mg g−1).30 Clearly, lignin exhibited an efficient and reliable adsorption capacity for Cr(VI).
 |
| | Fig. 5 The adsorption isotherms of Cr(VI) adsorbed onto lignin in aqueous solution (pH = 2.0, t = 2 h, adsorbent dose = 0.3 g L−1). | |
3.3.3 Trace Cr(VI) removal. The effect of the treatment time on the adsorption of trace Cr(VI) was performed to better illustrated its advantages, as compared to the direct electrochemical reduction mentioned above. As shown in Fig. 6, the adsorption rate was considerably rapid and attained equilibrium within 2 hours, suggesting a good electrostatic affinity between the negatively charged Cr(VI) ions and the protonated (positively charged) functional groups present on the lignin surface. More importantly, a nearly 94% adsorption removal of 2 ppm Cr(VI) was achieved as compared to the value of 22% via direct electroreduction at pH = 2.0, indicating lignin-based adsorption as an efficient and cost-effective approach for the removal of trace Cr(VI). The adsorption efficiency of Cr(VI) onto lignin was also measured by ICP-AES, and consistent results were obtained, showing that either UV-Vis spectroscopy or ICP-AES can be employed to effectively monitor the concentration of Cr(VI).
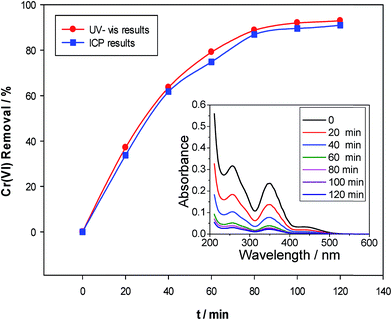 |
| | Fig. 6 Plots of the removal of Cr(VI) by lignin (pH = 2.0, lignin = 0.3 g L−1) versus the adsorption time determined using the UV-Vis spectroscopy (circle) and ICP-AES analysis. Inset: the comparison of UV-Vis spectroscopy of the solution prior to and following different treatment times. | |
3.4 Enhanced electrochemical Cr(VI) reduction by preconcentration
It is known that the electrochemical strategy for trace contaminant removal typically suffers from concentration-dependent slow mass transfer and sluggish reaction kinetics, leading to low current efficiency. As shown in the above section, the removal efficiency of electrochemical reduction for 2 ppm Cr(VI) was found to be only 22% following a 2 hour treatment period. In order to improve electrochemical performance, a lignin-mediated adsorption-release process was carried out. After two-hour adsorption treatment, ∼94% of 2 ppm Cr(VI) was efficiently removed due to a good electrostatic affinity between Cr(VI) ions and protonated functional groups on the lignin surface. To release and pre-concentrate the adsorbed Cr(VI), the NaOH solution was employed and the final pH was set to be 11, while its volume was 1/10 of the previous solution in the adsorption section. As shown in Fig. 7, the Cr(VI) concentration was found to be 18.8 ppm and a release efficiency of 100% was achieved, which is due to the fact that the adsorbent lignin could be completely dissolved in the alkaline solution.
 |
| | Fig. 7 (a) Cyclic voltammograms recorded at Ti/TiO2NT/Au electrode in the blank solution, 3 g L−1 lignin solution and the preconcentrate Cr(VI) solution (3 g L−1 lignin, 18.8 ppm Cr(VI)), pH = 11.0; (b) removal of Cr(VI) by electrochemical treatment at Ti/TiO2NT/Au electrode. | |
Fig. 7a presents the CVs of the Ti/TiO2NT/Au electrode recorded in the blank, lignin and lignin-mediated Cr(VI) solutions, respectively. The CV curve for the blank solution exhibited the hydrogen adsorption–desorption peaks at a potential of between −0.45 and −0.86 V, while there were two wide and broad reduction peaks that emerged at the potentials of ∼−0.12 and −0.70 V in the CV curve of lignin solution, which were possibly due to the adsorption of lignin and its reduction intermediate at the electrode surface.29 In contrast, there was a sharp and well defined reduction peak at −0.74 V, with its corresponding weak oxidation peak at −0.59 V, which was attributed to Cr(VI) reduction and re-oxidation. It was suggested that the lignin-related reductions were inhibited by the disappearance of the reduction reaction at −0.12 V, while the change of other at −0.70 V could not be distinguished. This was due to the fact that gold nanoparticle based electrodes are highly electrochemically reactive toward the reduction of Cr(VI).17 More importantly, a peak current of ∼350 μA for the reduction of Cr(VI) was observed in the preconcentrate solution, as compared to 44 μA in the original solution at 2 ppm Cr(VI). Clearly, the electrochemical Cr(VI) reduction was significantly improved due to the strategy of a lignin-mediated adsorption-release process.
To further confirm the enhanced performance of electrochemical Cr(VI) reduction, chronoamperometry was performed at a fixed potential of −0.74 V, which is the peak potential of Cr(VI) reduction (Fig. 7a). As shown in Fig. 7b, an initial current density of 402 μA cm−2 was observed, and a good current density stability was obtained. This was due to the lignin-mediated preconcentration and the rapid removal of the reaction product as Cr(III) precipitate (eqn (2)) in an alkaline solution. As a result, the electrochemical reduction efficiency of 18.8 ppm Cr(VI) was found to be 89% in the preconcentrate solution (Table 1). For comparison, chronoamperometry was also performed at −0.74 V in a 20 mL of 2 ppm Cr(VI) solution, where pH was adjusted to 11.0 using NaOH, revealing that the electrochemical reduction efficiency of Cr was 21.5% during the two-hour treatment period. The dramatic decrease of the electrochemical reduction efficiency can be attributed to the much slower of the mass transfer of Cr(VI) from the bulk solution to the electrode surface due to the significant decrease of the initial concentration of Cr(VI). In addition, the reaction product was insoluble Cr(OH)3, which could be easily recovered from the resulting suspension, indicating the integrated lignin-mediated adsorption-release process and electrochemical reduction is a promising practical application for the removal of trace Cr(VI).
Table 1 Comparison of the electrochemical performance for Cr(VI) reduction prior to and following the preconcentration step
| CCr(VI)/ppm |
Epca/V |
ipca/μA cm−2 |
isb/μA |
ifb/μA |
Removal/% |
pH |
| Peak potential and current density of Cr(VI) reduction in cyclic voltammetry curves. Initial (is) and final (if) current density of Cr(VI) reduction in chronoamperometry measurements. |
| 2 |
0.18 |
44 |
55 |
12 |
22 |
2.0 |
| 18.8 |
−0.74 |
350 |
402 |
360 |
89 |
11.0 |
4. Conclusions
A novel integrated lignin-mediated adsorption-release process and electrochemical reduction for the removal of trace Cr(VI) was developed. By adjusting the pH and volume of solutions, the lignin-mediated adsorption and release of Cr(VI) was achieved. Nearly 94% of 2 ppm Cr(VI) was removed by adsorption onto the surface of lignin in comparison to the value of 22% via direct electroreduction at Ti/TiO2NT/Au electrode in acidic solution. The Cr(VI) concentration was found to be 18.8 ppm subsequent to the 100% efficient release process, which was due to complete dissolution of the lignin in the alkaline solution. Moreover, the removal efficiency of Cr(VI) via electrochemical reduction was significantly improved to 89% in the preconcentrate solution. The reaction product was comprised of insoluble Cr(OH)3, which could be easily recovered from the suspension, showing that the new integrated lignin-mediated adsorption-release and electrochemical reduction processes proposed in this study is a promising strategy for the removal and recovery of trace concentrations of Cr(VI).
Acknowledgements
This work was supported by a Strategic Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC). A. Chen Acknowledges NSERC and the Canada Foundation of Innovation (CFI) for the Canada Research Chair Award in Material and Environmental Chemistry.
References
- S. E. Bailey, T. J. Olin and R. M. Brick, Water Res., 1999, 33, 2469–2479 CrossRef CAS.
- K. P. Lee, C. E. Ulrich, R. G. Geil and H. J. Trochimowicz, Sci. Total Environ., 1989, 86, 83–108 CrossRef CAS.
- J. Wang, K. Pan, E. P. Giannelis and B. Cao, RSC Adv., 2013, 3, 8978–8987 RSC.
- D. Rai, B. M. Sass and D. A. Moore, Inorg. Chem., 1987, 26, 345–349 CrossRef CAS.
- M. Cespon-Romero, M. C. Yebru.-Biurru and M. P. Bermejo-Barrera, Anal. Chim. Acta, 1996, 327, 37–45 CrossRef.
- J. Kotas and Z. Stasicka, Environ. Pollut., 2000, 107, 263–283 CrossRef CAS.
- W. Jin, S. Zheng, H. Du, H. Xu and Y. Zhang, Ind. Eng. Chem. Res., 2010, 49, 8244–8247 CrossRef CAS.
- Y. T. Lin and C. P. Huang, Sep. Purif. Technol., 2008, 63, 191–199 CrossRef CAS PubMed.
- P. Malaviya and A. Singh, Crit. Rev. Environ. Sci. Technol., 2011, 41, 1111–1172 CrossRef CAS.
- Y. Sag and T. Kutsal, Chem. Eng. J., 1995, 60, 181–188 CAS.
- Y. Zhang, Q. Li, R. Tang, Q. Hu, L. Sun and J. Zhai, Appl. Catal., B, 2009, 92, 351–356 CrossRef CAS.
- W. Jin, M. S. Moats, S. Zheng, H. Du, Y. Zhang and J. D. Miller, J. Phys. Chem. B, 2012, 116, 7531–7537 CrossRef CAS PubMed.
- M. G. Arroyo, V. Perez-Herrana, M. T. Montanes, J. Garcia-Anton and J. L. Guinon, J. Hazard. Mater., 2009, 169, 1127–1133 CrossRef CAS PubMed.
- I. Zongo, J.-P. Leclerc, H. A. Maiga, J. Wethe and F. Lapicque, Sep. Purif. Technol., 2009, 66, 159–166 CrossRef CAS PubMed.
- A. J. Chaudhary, N. C. Goswami and S. M. Grimes, J. Chem. Technol. Biotechnol., 2003, 78, 877–883 CrossRef CAS.
- H. A. Duarte, K. Jha and J. W. Weidner, J. Appl. Electrochem., 1998, 28, 811–817 CrossRef CAS.
- W. Jin, G. Wu and A. Chen, Analyst, 2014, 139, 235–241 RSC.
- S. A. Cavaco, S. Fernandes, M. M. Quina and L. M. Ferreira, J. Hazard. Mater., 2007, 144, 634–638 CrossRef CAS PubMed.
- L. Alvarado, I. R. Torres and A. Chen, Sep. Purif. Technol., 2013, 105, 55–62 CrossRef CAS PubMed.
- G. Pugazhenthi, S. Sachan, N. Kishore and A. Kumar, J. Membr. Sci., 2005, 254, 229–239 CrossRef CAS PubMed.
- L. Hsu, S. A. Masuda and K. H. Nealson, RSC Adv., 2012, 2, 5844–5855 RSC.
- A. S. K. Kumar and N. Rajesh, RSC Adv., 2013, 3, 2697–2709 RSC.
- J.-H. Chen, K.-C. Hsu and Y.-M. Chang, Ind. Eng. Chem. Res., 2013, 52, 11685–11694 CrossRef CAS.
- D. D. Das, R. Mahapatra, J. Pradhan, S. N. Das and R. S. Thakur, J. Colloid Interface Sci., 2000, 232, 235–240 CrossRef CAS PubMed.
- T. Karthikeyan, S. Rajgopal and L. R. Miranda, J. Hazard. Mater., 2005, B124, 192–199 CrossRef PubMed.
- M. Dakiky, M. Khamis, A. Manassra and M. Mereb, Adv. Environ. Res., 2002, 6, 533–540 CrossRef CAS.
- S. Hena, J. Hazard. Mater., 2010, 181, 474–479 CrossRef CAS PubMed.
- P. Miretzky and A. F. Cirelli, J. Hazard. Mater., 2010, 180, 1–19 CrossRef CAS PubMed.
- W. Jin, R. Tolba, J. Wen, K. Li and A. Chen, Electrochim. Acta, 2013, 107, 611–618 CrossRef CAS PubMed.
- A. B. Albadarin, A. H. Al-Muhtaseb, N. A. Al-laqtah, G. M. Walker, S. J. Allen and M. N. M. Ahmad, Chem. Eng. J., 2011, 169, 20–30 CrossRef CAS PubMed.
- A. Tejado, C. Pena, J. Labidi, J. M. Echeverria and I. Mondragon, Bioresour. Technol., 2007, 98, 1655–1663 CrossRef CAS PubMed.
- T. Dizhbite, G. Zakis, A. Kizima, E. Lazareva, G. Rossinskaya, V. Jurkjane and G. Telysheva, Bioresour. Technol., 1999, 67, 221–228 CrossRef.
- L. Dupont and E. Guillon, Environ. Sci. Technol., 2003, 37, 4235–4241 CrossRef CAS.
- C. P. Huang and W. H. Wu, Water Res., 1977, 11, 673–679 CrossRef CAS.
- X. Qu, M. Tian, B. Liao and A. Chen, Electrochim. Acta, 2010, 55, 5367–5374 CrossRef CAS PubMed.
- J. A. Laszlo, Environ. Technol., 1999, 20, 607–615 CrossRef CAS.
- M. Tian, M. Malig, S. Chen and A. Chen, Electrochem. Commun., 2011, 13, 370–373 CrossRef CAS PubMed.
- S. Chen, M. Malig, M. Tian and A. Chen, J. Phys. Chem. C, 2012, 116, 3298–3304 CAS.
- A. K. M. Kafi, G. Wu and A. Chen, Biosens. Bioelectron., 2008, 24, 566–571 CrossRef CAS PubMed.
- M. Tian, J. Wen, D. MacDonald, R. M. Asmussen and A. Chen, Electrochem. Commun., 2010, 12, 527–530 CrossRef CAS PubMed.
- J. Nam, N. Won, H. Jin, H. Chung and S. Kim, J. Am. Chem. Soc., 2009, 131, 13639–13645 CrossRef CAS PubMed.
- W. Jin, M. S. Moats, S. Zheng, H. Du, Y. Zhang and J. D. Miller, Electrochim. Acta, 2011, 56, 8311–8318 CrossRef CAS PubMed.
- B. Saha and C. Orvig, Coord. Chem. Rev., 2010, 254, 2959–2972 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 






