The pentafluorosulfanyl group in cannabinoid receptor ligands: synthesis and comparison with trifluoromethyl and tert-butyl analogues†
Stefano Altomontea,
Gemma L. Baillieb,
Ruth A. Rossb,
Jennifer Rileyc and
Matteo Zanda*ad
aKosterlitz Centre for Therapeutics, Institute of Medical Sciences and “John Mallard” Scottish PET Centre, University of Aberdeen, Foresterhill, Aberdeen AB25 2ZD, Scotland, UK. E-mail: m.zanda@abdn.ac.uk
bMedical Sciences Building, University of Toronto, 1 King's College Circle, Toronto, Ontario M5S 1A8, Canada
cDrug Discovery Unit, University of Dundee, Dow Street, Dundee, DD1 5EH, Scotland, UK
dC.N.R.-Istituto di Chimica del Riconoscimento Molecolare, via Mancinelli 7, 20131 Milano, Italy. Fax: +44 01224 437465
First published on 15th April 2014
Abstract
An array of cannabinoid ligands, bearing meta- and para-substituted pentafluorosulfanyl (SF5) aniline groups in position 3 of the pyrazole ring, was efficiently synthesised and compared with the exact trifluoromethyl and tert-butyl analogues. In general, the SF5 substituted ligands showed higher lipophilicity (i.e. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values) than the CF3 counterparts and lower lipophilicity than the tert-butyl ones. In terms of pharmacological activity, SF5 pyrazoles generally showed slightly higher or equivalent CB1 receptor affinity (Ki), always in the nanomolar range, and selectivity towards the CB2 relative to both CF3 and tert-butyl analogues. Functional β-arrestin recruitment assays were used to determine equilibrium dissociation constants (Kb) and showed that all of the tested SF5 and CF3 compounds are CB1 neutral antagonists. These results confirm the possibility of successfully using an aromatic SF5 group as a stable, synthetically accessible and effective bioisosteric analogue of the electron-withdrawing CF3 group, and possibly also of bulky aliphatic groups, for drug discovery and development applications.
P values) than the CF3 counterparts and lower lipophilicity than the tert-butyl ones. In terms of pharmacological activity, SF5 pyrazoles generally showed slightly higher or equivalent CB1 receptor affinity (Ki), always in the nanomolar range, and selectivity towards the CB2 relative to both CF3 and tert-butyl analogues. Functional β-arrestin recruitment assays were used to determine equilibrium dissociation constants (Kb) and showed that all of the tested SF5 and CF3 compounds are CB1 neutral antagonists. These results confirm the possibility of successfully using an aromatic SF5 group as a stable, synthetically accessible and effective bioisosteric analogue of the electron-withdrawing CF3 group, and possibly also of bulky aliphatic groups, for drug discovery and development applications.
Introduction
Pentafluorosulfanyl group
Although only a few fluorinated natural compounds have been isolated,1 it is well known that introduction of one or more fluorine atoms into a molecule can have profound effects on the binding to a receptor, and can improve both its metabolic stability and bioavailability.2 Fluorine could be incorporated by fluorination or alternatively via a building-block approach. Among the fluorinated motifs, the trifluoromethyl group occupies a prominent role in drug discovery,3 and several blockbuster drugs display a CF3 substituent.4In 1960 Sheppard reported for the first time the synthesis of an aromatic compound bearing a pentafluorosulfanyl group, SF5.5 However, due to of the inconvenient synthetic access to pentafluorosulfanyl arenes, the breakthrough in the commercialization first and then in the application of SF5-compounds in drug discovery and materials science came in the late 90s, with the improvement of their synthesis.
The synthesis of pentafluorosulfanyl compounds, their biological applications and properties have been reviewed.6–9 Importantly, the pentafluorosulfanyl group is often compared to the trifluoromethyl group, and because of its higher lipophilicity,10 electronegativity,11 chemical stability and greater steric demand, which is only slightly lower than that of the tert-butyl group,12 the SF5 group is often referred to as a “super-trifluoromethyl” group (Fig. 1).13–15
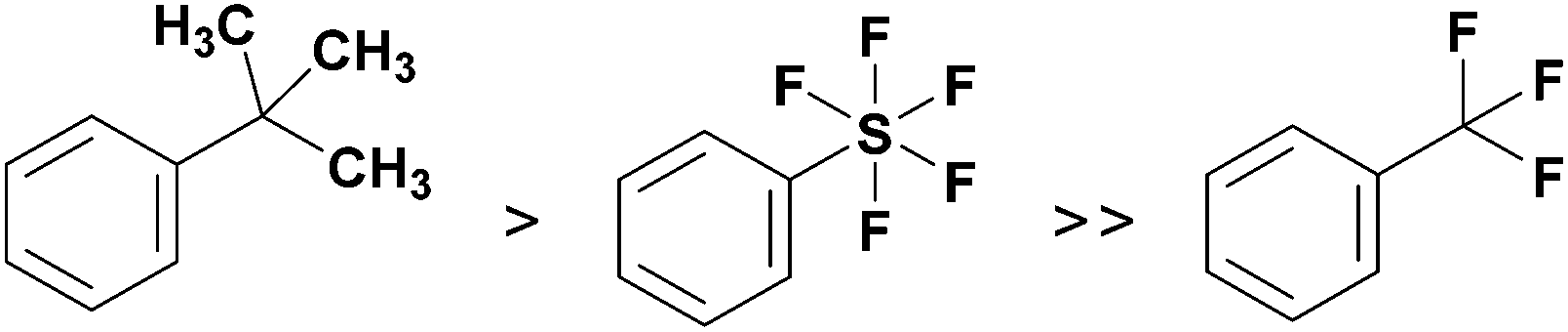 | ||
| Fig. 1 Steric demand of the three groups compared in this work: tBu > SF5 ≫ CF3. | ||
Cannabinoid receptors
Cannabinoid receptors belong to the G-protein coupled receptors family (GPCRs).16,17 At least two cannabinoid receptor subtypes have been identified: CB1 and CB2,18 furthermore, the CB1 type has two splice variants, denominated CB1A and CB1B.19,20 The distribution of CB1 receptors is localised predominantly in the brain21 whereas the CB2 are present in the peripheral nervous system (PNS) cells.22 However, recent studies have demonstrated the presence of CB1 in the PNS23 and, on the other hand, of the CB2 in the central nervous system, albeit in low density.24 Since CB1 receptors are associated with several disorders, such as depression,25 anxiety,26 stress,27 schizophrenia,28 chronic pain29 and obesity,30 several cannabinoid ligands were developed. Among these ligands, the most studied is probably SR141716 (Rimonabant),31 a pyrazole-core inverse agonist which was discovered by Sanofi-Synthelabo (now Sanofi-Aventis) in 1994 (Fig. 2), marketed in Europe as anti-obesity drug and subsequently withdrawn from the market owing to its side-effects.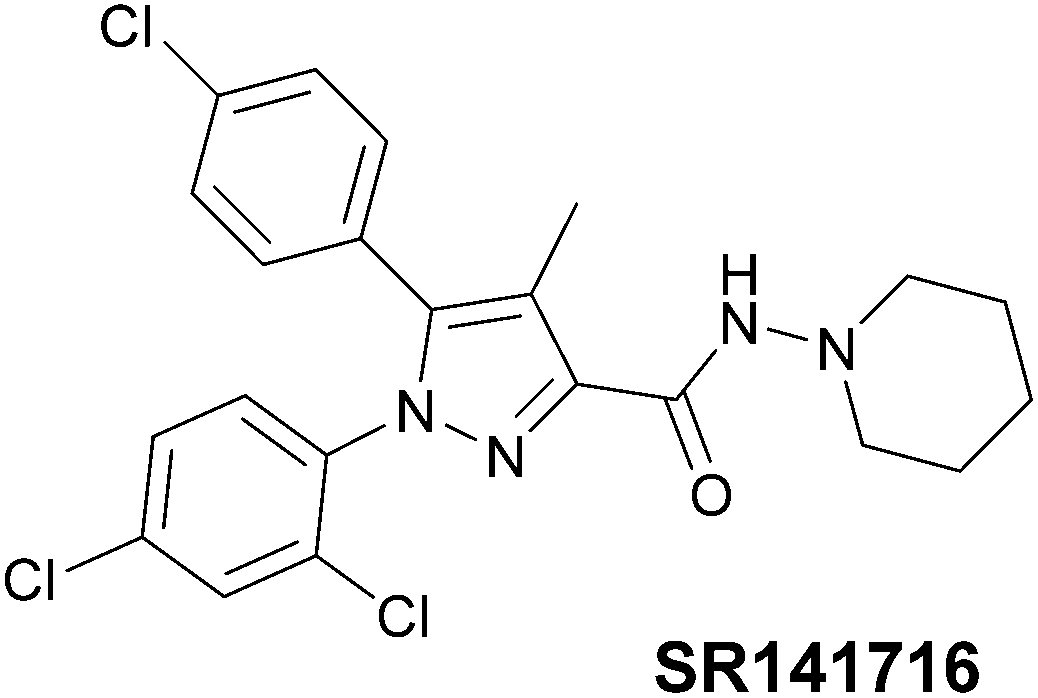 | ||
| Fig. 2 Chemical structure of Rimonabant (SR141716). | ||
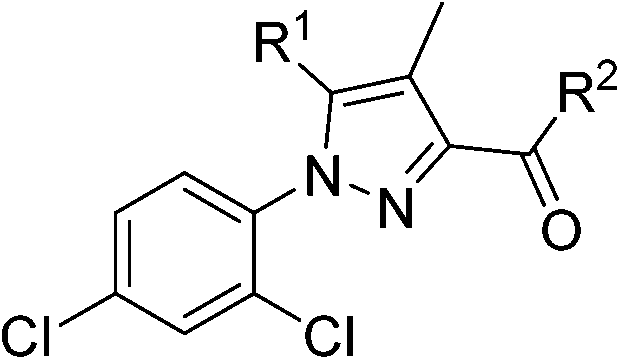
The scientific question we wanted to address in this work was about the position occupied by the SF5 group relative to its closest bioisosteric substituents, namely CF3 and tert-butyl groups, in terms of its effect on key pharmacological and physico-chemical properties, such as lipophilicity and solubility. To answer, we decided to use a Rimonabant-type scaffold as a model bioactive structure for incorporating an SF5-group, as well as CF3 and the tert-butyl groups, and directly compare the SF5 derivatives with their CF3 and tert-Bu counterparts from the pharmacological viewpoint. SF5, CF3 or tert-butyl groups were incorporated on a carboxy-aniline residue in position 3 of the pyrazole ring, since (1) 3-carboxy-aniline Rimonabant analogues were shown to have excellent CB1-affinity and selectivity vs. the CB232 and (2) SF5-substituted anilines or nitro-anilines are accessible starting materials (see below).
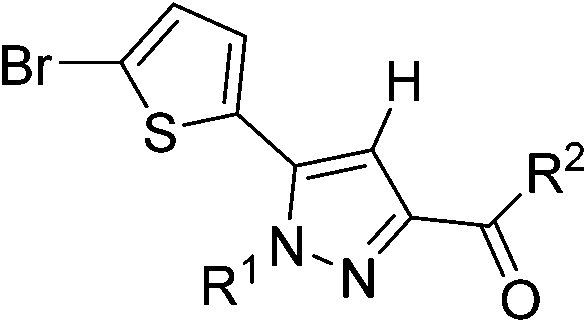
Since, to the best of our knowledge, SF5-substituted cannabinoid receptor ligands have never been described in the literature, we decided to synthesise two different classes of pyrazole-core CB1 receptor ligands: the former based on a Rimonabant-type structure and the latter based on ligands described in a Pharmaness' patent,33 where the 4-chloro-phenyl ring is replaced by a 2-bromo-thiophene.
Results and discussion
Chemistry
The starting para-SF5-substituted aniline is commercially available whereas meta-SF5-aniline was synthesised from the commercially available 3-nitro-SF5 benzene (Scheme 1). 3-Nitro-SF5-benzene was treated with iron-powder in a refluxing HCl–ethanol solution,34 affording the corresponding 3-(pentafluoro-λ6-sulfanyl)aniline in good yield (83%). The SF5 group remained unreactive under these conditions, confirming the high chemical stability of this group.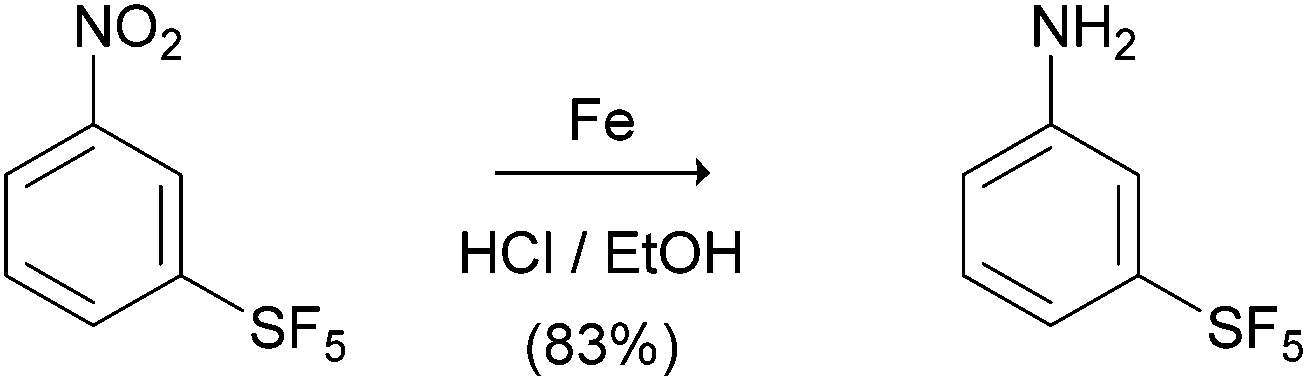 | ||
| Scheme 1 Reagents and conditions: Fe, HCl–EtOH, reflux, 2 h. | ||
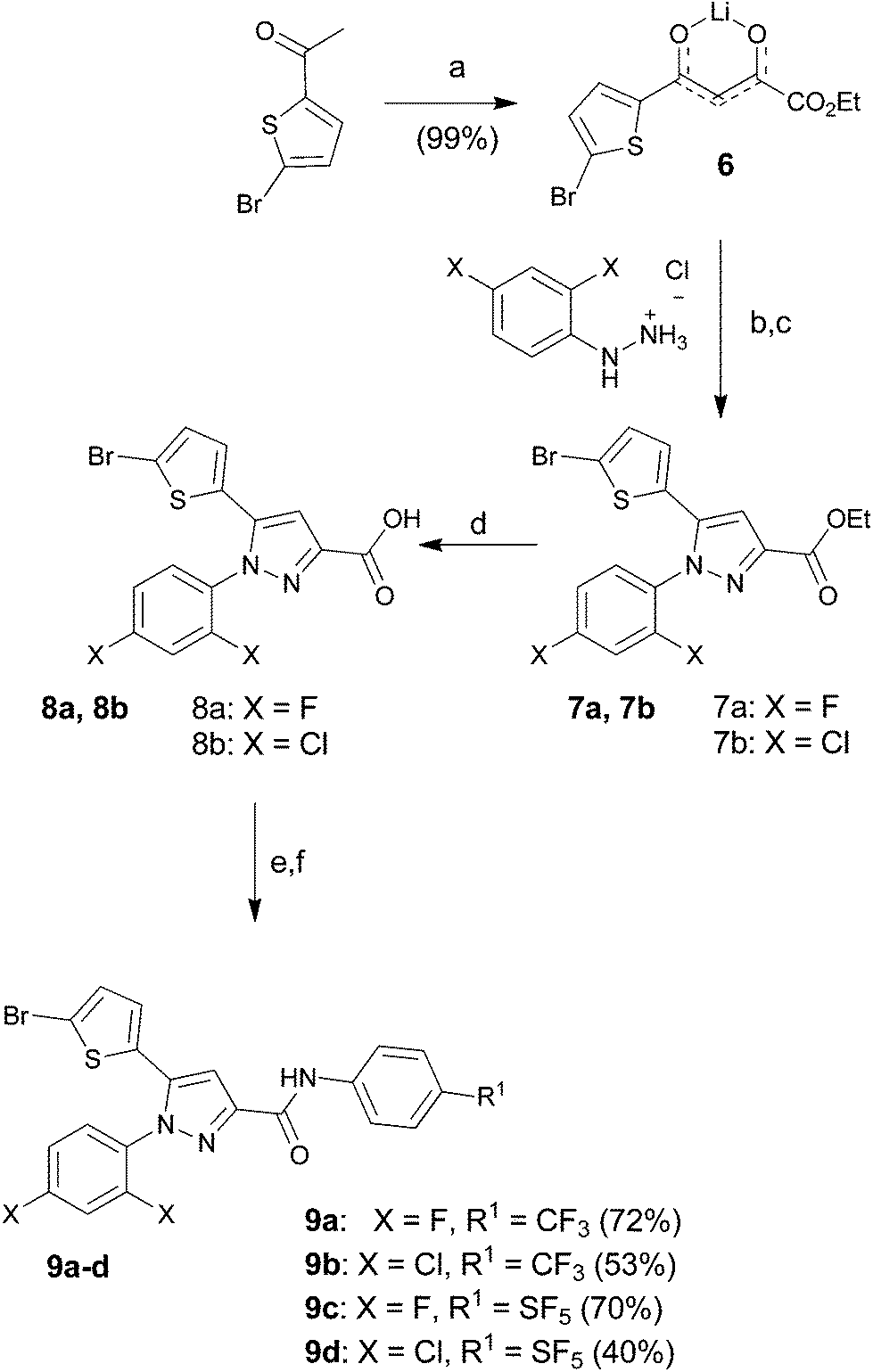
Thiophenyl-compounds 5a–f and 9a–d were prepared according to the general synthetic methods shown in Schemes 2 and 3, respectively.35 4-Methyl-pyrazole-substituted compounds 5 were synthesised starting with a reaction of 2-propionyl-thiophene with diethyl oxalate using LHMDS as a base which provided the stable lithium salt 1 in moderate yields. The latter was allowed to react first with 2,4-dihalo-phenylhydrazine hydrochloride in ethanol, followed by intramolecular cyclization in refluxing acetic acid to provide the pyrazole ester 2. Treatment of 2 with NBS afforded the 2-bromothiophene 3 in 90% yield via regioselective bromination in position 5 of the thiophene ring. The ester 3 was hydrolysed under basic conditions to give the carboxylic acid 4 in very good yield. The acid 4 was first converted into the corresponding acyl chloride with thionyl chloride and then reacted with several anilines to afford the desired products 5.
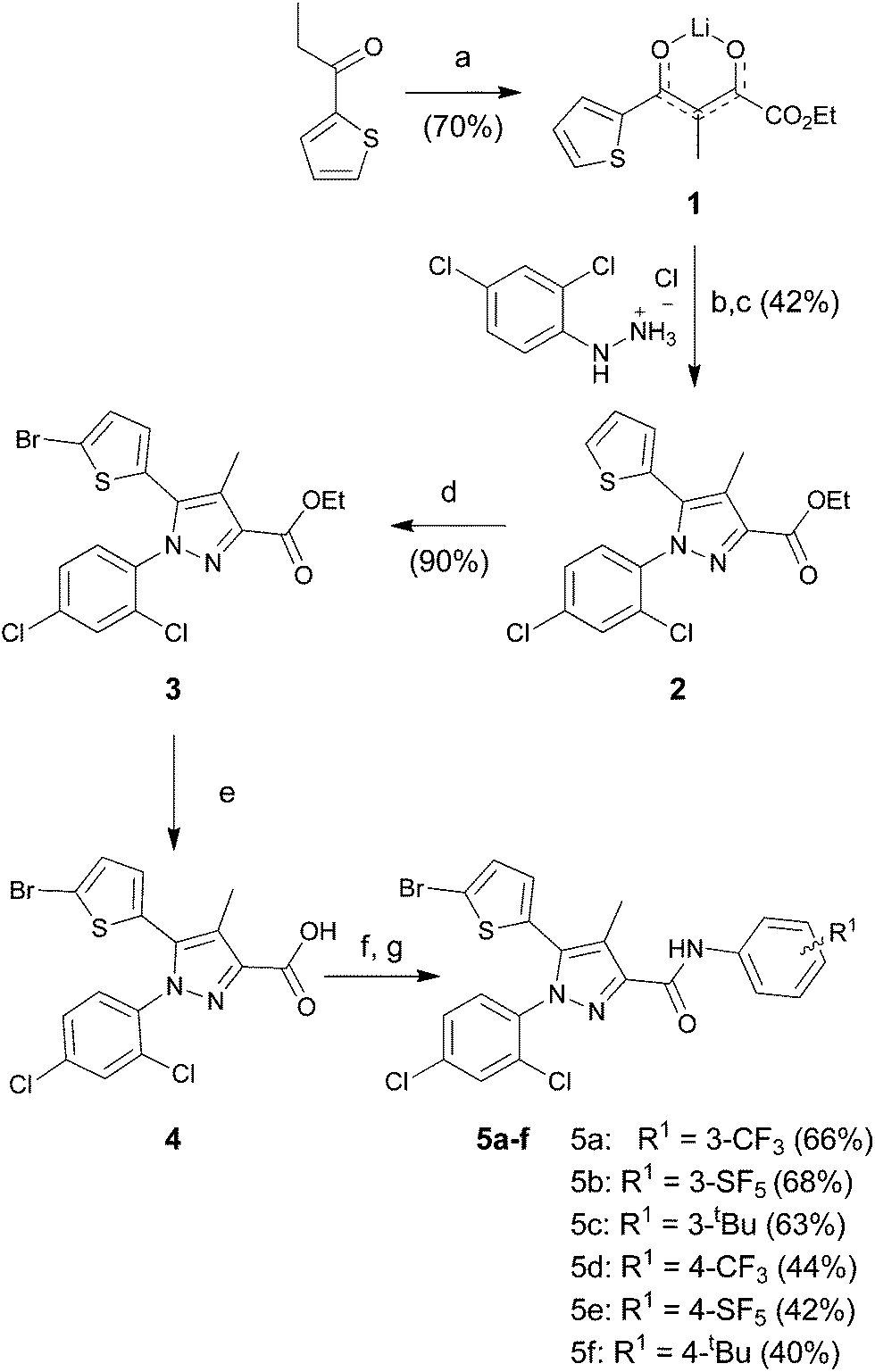 | ||
| Scheme 2 Reagents and conditions: (a) diethyl oxalate, LiHMDS, THF/Et2O (2/1), −78 °C to room temp., 16 h; (b) EtOH, room temp., 24 h; (c) AcOH, 120 °C, 16 h; (d) NBS, CH3CN, 0 °C to r.t., 16 h; (e) KOH, MeOH, reflux, 3 h; (f) thionyl chloride, toluene, reflux, 3 h; (g) Et3N, DCM, 0 °C to r.t., 16 h. | ||
 | ||
| Scheme 3 Reagents and conditions: (a) diethyl oxalate, LiHMDS, THF/Et2O (2/1), −78 °C to room temp, 16 h; (b) EtOH, room temp, 24 h; (c) AcOH, 120 °C, 16 h; (d) KOH, MeOH, reflux, 3 h; (e) thionyl chloride, toluene, reflux, 3 h; (f) Et3N, DCM, 0 °C to room temp, 16 h. | ||
Similar procedure was used for the synthesis of the pyrazoles 9 (Scheme 3), having no substitution in position 4. In this case, however, the overall synthesis was one-step shorter thanks to the commercial availability of 2-bromo-5-acetyl-thiophene, which allowed us to skip the bromination reaction.
The synthesis of the Rimonabant-like derivatives 13, shown in Scheme 4, was analogously accomplished following the strategy described by Lan et al.32
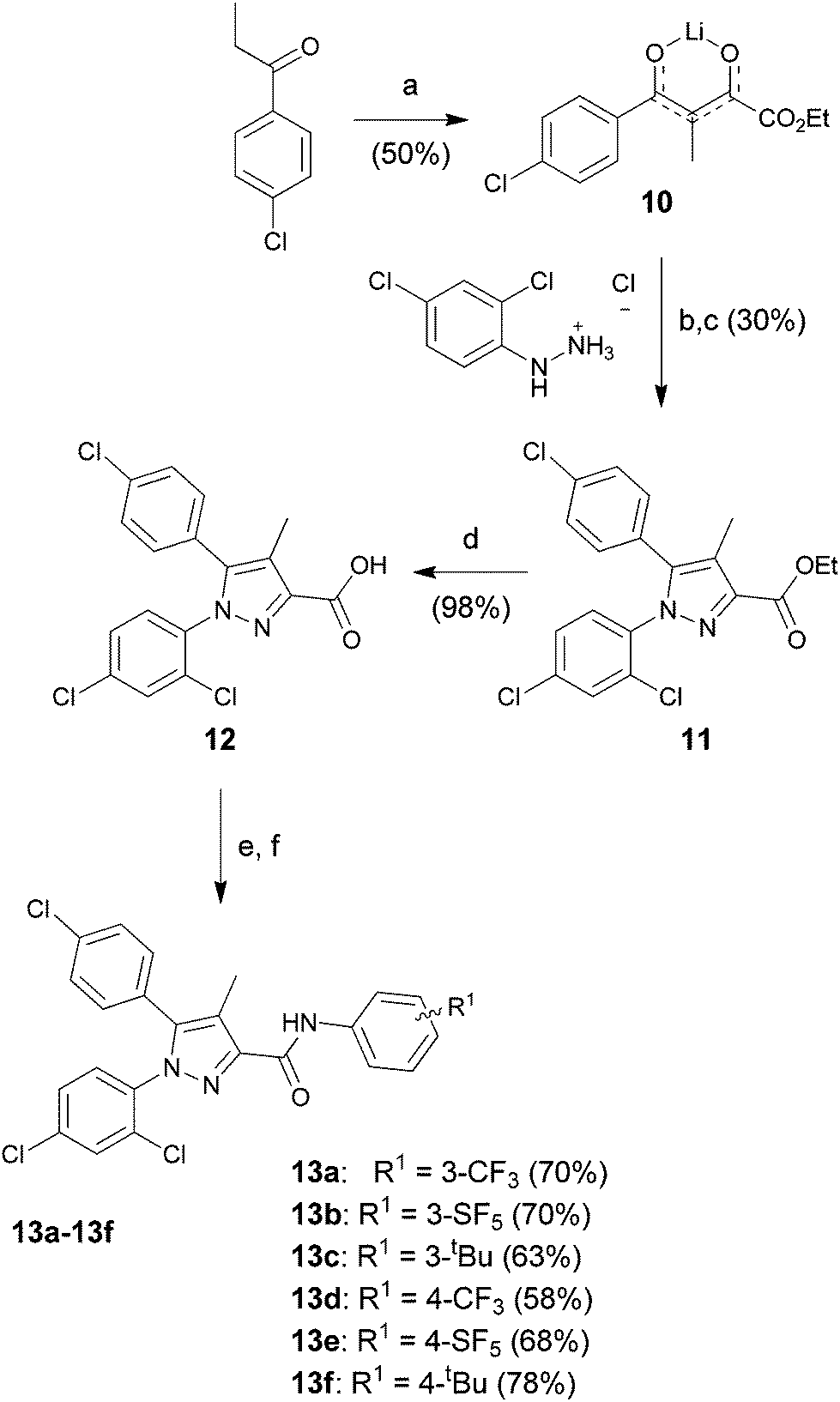 | ||
| Scheme 4 Reagents and conditions: (a) diethyl oxalate, LiHMDS, THF/Et2O (2/1), −78 °C to room temp, 16 h; (b) EtOH, room temp, 24 h; (c) AcOH, 120 °C, 16 h; (d) KOH, MeOH, reflux, 3 h; (e) thionyl chloride, toluene, reflux, 3 h; (f) Et3N, DCM, 0 °C to room temp, 16 h. | ||
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P measurement
P measurement
Streich et al. showed that an SF5 substituent on phenoxyacetic acid imparts higher lipophilicity than a CF3 group.10 We therefore investigated whether this was the case for our pyrazole cannabinoid ligands too.
Several software packages allow the prediction of physicochemical molecular properties, including the logP (octanol/water partition coefficient). However, there are often significant discrepancies among the calculated values. We therefore decided to set up an experimental method for determining the logP of these CB1 ligands via reverse phase-HPLC analysis.
The retention times obtained (which are proportional to the lipophilic character of the molecules) confirmed that molecules bearing the SF5 moiety are more lipophilic than the CF3 counterparts and, in general, a substituent in para position influences the hydrophobicity of the entire molecule to a greater extent (ΔlogP(SF5–CF3)p = 0.3) than in meta (ΔlogP(SF5–CF3)m = 0.2) (Tables 1 and 2). Not surprisingly, replacement of these fluorinated functions with a strongly lipophilic tert-butyl group further increased the logP of the molecules.
|
|||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | logPa (±SEM) |
CB1 Ki (nM) (±SEM) | CB2 Ki (nM) (±SEM) |
| a Determined experimentally by means of RP-HPLC (see experimental section for details). | |||||
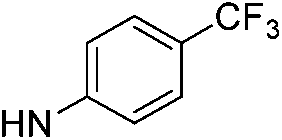
| 9a | 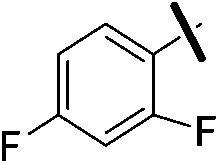 |
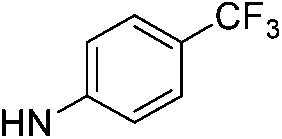 |
4.67 (±0.20) | 817.8 (±247.2) | 2991.0 (±988.2) |
| 9b | 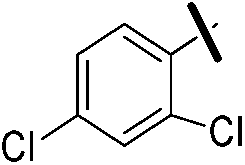 |
 |
5.60 (±0.27) | 137.7 (±36.9) | 528.4 (±204.8) |
| 9c | 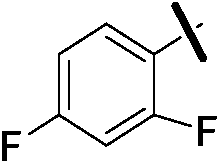 |
 |
4.97 (±0.22) | 302.7 (±72.1) | 1009.0 (±349.2) |
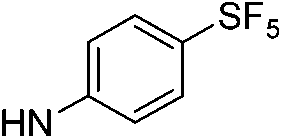
| 9d |  |
 |
5.89 (±0.30) | 58.8 (±13.4) | 588.2 (±229.9) |
|
|||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | logPa (±SEM) |
CB1 Ki (nM) (±SEM) | CB2 Ki (nM) (±SEM) |
| a Determined experimentally by means of RP-HPLC (see Experimental section for details). | |||||
| 5a |  |
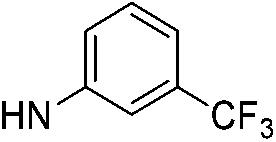 |
6.19 (±0.32) | 50.9 (±6.1) | 362.2 (±67.6) |
| 5d | 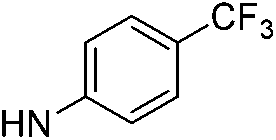 |
6.34 (±0.34) | 65.3 (±13.0) | 706.6 (±309.7) | |
| 5b | 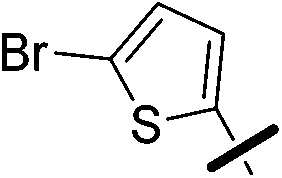 |
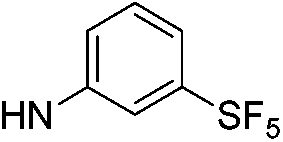 |
6.42 (±0.35) | 66.1 (±15.5) | 991.2 (±231.7) |
| 5e | 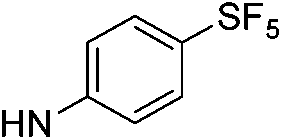 |
6.61 (±0.37) | 7.9 (±3.3) | 306.4 (±140.3) | |
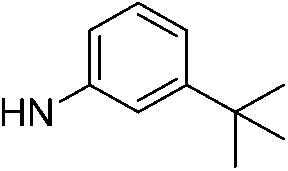
| 5c | 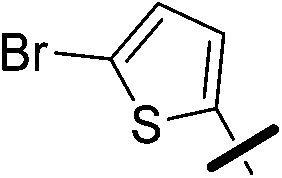 |
 |
7.09 (±0.40) | 19.9 (±4.6) | 102.1 (±25.0) |
| 5f | 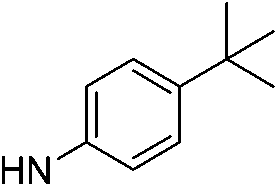 |
7.04 (±0.41) | 106.9 (±30.1) | 906.2 (±361.0) | |
| 13a | 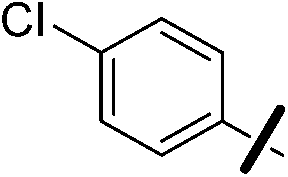 |
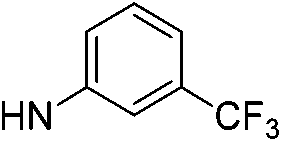 |
5.97 (±0.30) | 26.5 (±6.4) | 609.6 (±195.5) |
| 13d | 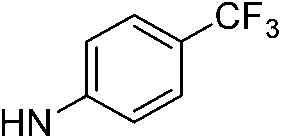 |
6.14 (±0.32) | 31.3 (±9.8) | 392.8 (±91.4) | |
| 13b | 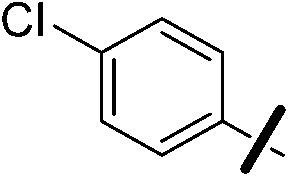 |
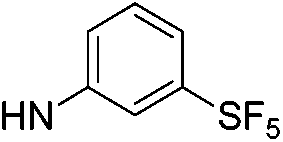 |
6.21 (±0.33) | 11.2 (±2.4) | 637.3 (±205.2) |
| 13e | 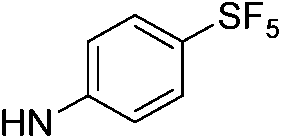 |
6.41 (±0.35) | 17.1 (±3.4) | 537.2 (±210.3) | |
| 13c | 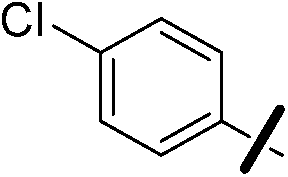 |
 |
6.95 (±0.38) | 8.5 (±1.5) | 1950.0 (±852.6) |
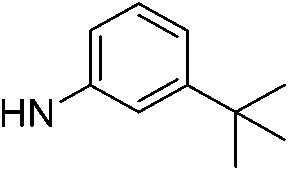
| 13f |  |
6.81 (±0.40) | 47.9 (±19.8) | 2053.0 (±949.8) | |
Replacement of the 4-methyl group on the pyrazole, as in compounds 5 and 13, with a hydrogen, as in compounds 9, reduced the lipophilicity of both SF5 and CF3 derivatives by ΔlogP(CH3–H)SF5 = 0.7 and ΔlogP(CH3–H)CF3 = 0.6 units respectively. A further hydrophilicity enhancement was observed by substitution of the 2,4 difluoro aryl group in 9a,c with the chlorinated analogue in 9b,d (ΔlogP(F–Cl) = 0.9).
When a 4-chloro-phenyl moiety (compounds 13) was replaced by a 5-bromo-thiophene group (compounds 5), the logP decreased by ΔlogP(Thy–Ph) = 0.2, despite the benzene group (logP benzene = 2.15)36 is reported to be more hydrophobic than the thiophene group (logP thiophene = 1.81).36 These results are presumably influenced by the presence on the thiophene ring of the bromine atom, which is softer and more hydrophobic than chlorine.
Most of the tabulated logP values for the CB1 inverse agonist Rimonabant (SR141716) were determined in silico, and the two experimental values reported in literature, obtained through the flask-shake technique, are quite different (logD7.4 = 4.6 ± 0.837 and logD7.4 = 3.838). In order to obtain a new experimental value we decided to test SR141716 using the previously described RP-HPLC method, which provided a logP value of 4.73 ± 0.20 for Rimonabant. This confirmed that all the new ligands presented in this article exhibit higher hydrophobicity than SR141716.
Binding affinity and SAR
Compounds 9a–d lack a substituent in position 4 of the pyrazole ring, incorporate a (5-bromo-thiophene) residue in position 5 and carry different aryl residues in position 1 (2,4-difluorophenyl for 9a,c and 2,4-dichlorophenyl for 9b,d). In this series, we compared the para-phenyl substituted compounds 9a and 9b, bearing a CF3 group, with, respectively, 9c and 9d, carrying an SF5 group (Table 1). In both cases, the SF5 compounds 9c,d showed higher affinity (lower Ki) than the CF3 counterparts. In terms of CB1/CB2 selectivity, while 9a,c were comparable, 9d showed modest but higher CB1/CB2 selectivity (10-fold) than 9b (4-fold).
It has been previously demonstrated that an aliphatic substituent in position 4 of the pyrazole ring imparts higher CB1 selectivity in pyrazole-based ligands; in particular Chen et al. performed a 3D quantitative structure–activity relationship (QSAR) of 5-aryl pyrazole structures using the comparative molecular field analysis (CoMFA),40 highlighting the importance of the 4-methyl group on pyrazole ring to achieve a better CB1/CB2 ratio.
We therefore investigated also the effect of SF5, CF3 and tert-butyl groups as 3-phenyl-carboxamide substituents in two series of 4-methyl-pyrazole cannabinoid ligands: 5-(5-bromo-thiophene)-pyrazoles 5a–f and Rimonabant-type 5-(4-chlorophenyl)-pyrazoles 13a–f.
As expected, the lower apparent Kis of all compounds 5 and 13 (Table 2) relative to 9a–d confirmed that introduction of a methyl group in position 4 of the pyrazole ring results in an affinity increase versus CB1 and, on the other hand, decreased the Emax measured by the displacement assay on CB2 CHO cells (see Table S1, ESI†).
In the para-phenyl substituted series of compounds 5d–f, the SF5-compound 5e showed the highest CB1 affinity, whereas the presence of the tert-butyl group in 5f caused a substantial drop both in affinity and CB1/CB2 selectivity. Interestingly, the CB1/CB2 selectivity was higher for the SF5 derivative 5e relative to the CF3 analogue 5d. In the meta-substituted series of compounds 5a–c, the tert-butyl derivative 5c featured the highest CB1 affinity, whereas the SF5 compound 5b and the CF3-analogue 5a showed comparable CB1 affinities.
For the 4-chlorophenyl Rimonabant-type series of compounds 13a–f (Table 2), we also observed nanomolar affinities for the CB1, in the same range of compounds 5a–f, but the CB1/CB2 selectivity was generally higher. In the meta-substituted series of compounds 13a–c, the SF5 derivative 13b and the tert-butyl 13c displayed the highest CB1 affinity, and 13b showed 2-fold higher affinity than the CF3 analogue. The situation was somewhat reversed for the para-substituted compounds 13d–f where the SF5 compound 13e displayed the lowest apparent CB1 Ki, whereas 13d and 13f showed similar affinities.
However, importantly, all of the 4-methyl-substituted pyrazoles bearing the SF5 or tBu groups in para position, namely 5e, 5f, 9d, 9c and 13f, displaced [3H]CP55940 only partially at the maximum concentration of 10−5 M with Emax values ranging from 35.3 to 43.2 (see Fig. 3 and Table S1, ESI†). Considering that the 4-trifluomethyl arene analogue 5d produced a nearly full displacement of [3H]CP55940, we initially hypothesised that this behaviour could be explained by the binding of the compound to a topographically distinct site on CB1 (an ‘allosteric binding site’). We therefore investigated the effect of thiophenyl 5e on the dissociation of [3H]CP55940 from mouse brain membranes; as this is the gold standard method of assessing an allosteric interaction.41 However, compound 5e had no significant effects on CB1 agonist dissociation indicating that it is not an allosteric modulator.
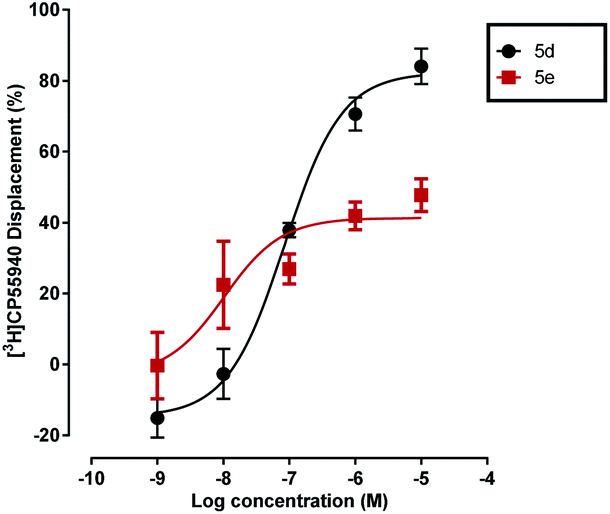 | ||
| Fig. 3 Effect of compounds 5d,e on equilibrium binding of [3H]CP55940 to mouse brain membranes. Data shown are mean ± SEM 3–4 independent experiments. Data were best fitted by a sigmoidal concentration–response curve. | ||
At that point, we hypothesised that the partial displacement might be due to a solubility issue at the highest concentrations tested, i.e. 10−5 M. In this context, Jackson et al. had previously shown that, although the S–F bond is longer and more polarizable than C–F one, the entropic cost that derives from dissolving in water a larger group played a major role, resulting in lower Sw (water solubility) values for most of the pentafluorosulfanyl analytes relative to their CF3 analogues.42 Laser nephelometry has become the method of choice for measuring solubility of molecules in a drug discovery setting.43
We therefore submitted compounds 5e, 5d and 5b to a laser nephelometry assay, for assessing the solubility of each compound at different concentrations. However, these experiments showed that all of the compounds above were soluble at the highest concentration of 10−5 M (99.9% aqueous buffer, 0.1% DMSO) (see Table S2, ESI,† for details).
Using the PathHunter® β-arrestin assay in hCB1 cells, [3H]CP55940 stimulated β-arrestin recruitment with an EC50 of 16.9 nM (11–27 nM) (95% confidence limits for all of the β-arrestin experiments herein described). The fluorinated para-substituted CB1 receptor ligands, 5e, 5d, 13e and 13d (Table 2) caused a significant rightward shift in the dose response curve of [3H]CP55940, with an EC50 of 111.0 nM (82–151 nM), 235.8 nM (157–355 nM), 245.8 nM (194–311 nM) and 439.6 nM (298–649 nM) respectively (see ESI† for the graphics). Analogously, with the fluorinated meta-substituted CB1 receptor ligands 13b, 13a, 5b, and 5a, [3H]CP55940 stimulated beta-arrestin recruitment with an EC50 of 17.2 nM (14–22 nM) and the SF5 compounds again, caused a significant rightward shift in the dose response curve of [3H]CP55940 with EC50 values of 932.6 nM (451–1927 nM), 1528 nM (336–6953 nM), 158.6 nM (116–218 nM) and 57.0 nM (42–77 nM), respectively.
All of the tested compounds produced a significant increase in the EC50 values with a rank order of efficacy of 13a, 13b, 13d, 13e, 5d, 5b, 5e, and 5a, and should be therefore considered competitive antagonists of [3H]CP55940 for the CB1 receptor.
Summary and conclusions
We have shown that the pentafluorosulfanyl group can effectively replace a trifluoromethyl group in pyrazole-type cannabinoid ligands. The resulting SF5-compounds behaved as competitive CB1 receptor antagonists, which is an interesting property since CB1 inverse agonists have been reported to produce severe adverse effects that limit their clinical applications, whereas CB1 neutral antagonists might have improved clinical utility.45 Furthermore SF5-compounds showed nanomolar inhibition (Ki) and equilibrium dissociation constants (Kb) for the CB1 receptor, with moderate CB1/CB2 selectivity.Both affinity and selectivity of SF5-compounds were generally slightly superior or comparable to that of CF3 and tert-butyl analogues. Experimental lipophilicity (logP) was found to follow the trend tert-butyl > SF5 > CF3. Functional assays showed that all of the tested compounds, belonging to the SF5 and CF3 series, are CB1 neutral antagonists. It is worth noting that some of the compounds, incorporating para-SF5- or tert-butyl-aryl groups on the C-3 pyrazole ring, displayed an apparent partial displacement of [3H]CP55940 in the functional assays (Emax values ranging from 35.3 to 43.2), while ligands binding to the orthosteric binding pocket would be expected to fully displace the radioligand. One explanation for this observation could be that the compounds are binding to an allosteric pocket. However, this seems unlikely as we observed no change in agonist dissociation. Solubility problems were also excluded by means of nephelometry assays, so we are currently unclear as to the explanation for this observation. Overall, the data collected in this work confirm that (1) an aromatic SF5 group is an effective bioisosteric analogue of the CF3 group, and possibly also of bulky aliphatic groups like the tert-butyl, (2) it can be successfully used as a substitutent in biologically active compounds and drug candidates.
Experimental section
Chemistry
NMR data were recorded on Bruker ADVANCE III for 1H at 400.13 MHz, for 13C at 100.58 MHz and for 19F at 376.45 MHz. 1H NMR chemical shifts are reported relative to TMS, and the solvent resonance was employed as the internal standard (CDCl3 δ = 7.26). 13C NMR spectra were recorded with complete proton decoupling, and the chemical shifts are reported relative to TMS with the solvent resonance as internal standard (CDCl3 δ = 77.0). 19F NMR spectra were referenced to CFCl3 as the external standard. All chemical shift (δ) are reported in parts per million (ppm) downfield from TMS and coupling constant (J) in Hertz. Splitting patterns are reported as follows: s, singlet; d, doublet; dd, doublet of doublets; ddd, doublet of doublet of doublets; t, triplet; td, triplet of doublets; appt, apparent triplet; q, quadruplet; qd, quadruplet of doublets; m, multiplet; br, broad signal.
Mass Analysis was performed using an Agilent 1200 HPLC system coupled to an Agilent G6120 single quadrupole detector equipped with electrospray ionization (ESI) source in direct infusion modality.
Lipophilicities were determined using a Reverse Phase (RP)-HPLC with an Agilent 1200 HPLC system equipped with a DAD, analytical Phenomenex Luna C-18 column (250 × 4.60 mm L × ID, particle size: 5 μ) and an ESI-MS detector.
HRMS analysis were performed using an LTQ Orbitrap XL MS spectrometer.
All reactions were carried out in oven- or flame-dried glassware under nitrogen atmosphere, unless stated otherwise, and were magnetically stirred and monitored by TLC on silica gel (60 F254 pre-coated glass plates, 0.25 mm thickness).
Visualization was accomplished using irradiation with a UV lamp (λ = 254 nm or λ = 365 nm), and/or staining with potassium permanganate or ceric ammonium molybdate solution.
Purification of reaction products was performed using flash chromatography on silica gel (60 Å, particle size 40–63 μm) according to the procedure of Still and co-workers.46
Yields refer to chromatographically and spectroscopically pure compounds, unless stated otherwise.
3-(Pentafluoro-λ6-sulfanyl)aniline. To a stirred acidic warm solution (50 °C) of pentafluoro(3-nitrophenyl)-λ6-sulfane (1.00 g, 3.93 mmol) in ethanol–HCl (11 mL:0.8 mL, 37% v/v), iron powder (1.22 g, 21.63 mmol) was added portionwise. The reaction was refluxed for 2 h. After cooling, the solid was removed by means of filtration; the filtered was diluted with dichloromethane. The organic phase was acidified with HCl 2N. The aqueous phase was separated, basified and extracted with dichloromethane (3 × 50 mL). The combined extracts were washed with brine, dried over anhydrous sodium sulfate, filtered, and evaporated to give the crude aniline as a yellow oil (0.72 g, 83% yield): the obtained 1H, 12C and 19F NMR spectra matched to those reported by Bowden et al.34
Lithium salt of ethyl 3-methyl-2,4-dioxo-4-(thiophen-2-yl)butanoate (1). To a magnetically stirred solution of lithium bis(tri-methylsilyl)amide (61 mL, 60.60 mmol, 1.0 M in THF) in diethyl ether (110 mL) at −78 °C, 1-(2-thienyl)-1-propanone (7 mL, 55.10 mmol) in diethyl ether (42 mL) was added dropwise under a nitrogen atmosphere. After the mixture was stirred at the same temperature for an additional 45 min, diethyl oxalate (9 mL, 66.11 mmol) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for another 16 h. The reaction precipitate was filtered, washed with diethyl ether, and dried under vacuum to afford the crude lithium salt 1 (9.50 g, 70%) as a pale-yellow solid. The product was used without further purification. ESMS, calculated m/z C11H11LiO4S 246.05 [M]+, found m/z (relative intensity) 247.0 [M + H]+ (100), 265.0 [M + H + Na − Li]+ (100), 279.0 [M + H + K − Li]+ (100).
1-(2,4-Dichlorophenyl)-4-methyl-5-thiophen-2-yl-1H-pyrazole-3-carboxylic acid ethyl ester (2). To a solution of lithium salt 1 (8.50 g, 33.83 mmol) in ethanol (26 mL) was added 2,4-dichlorophenylhydrazine hydrochloride (8.85 g, 40.60 mmol) in one portion at room temperature under nitrogen. The resulting mixture was stirred at the same temperature for 24 h. After the reaction was complete, the precipitate was filtered, washed with ethanol and diethyl ether, and dried under vacuum to give a light-yellow solid (7.47 g). This crude solid, without purification, was dissolved in acetic acid (68 mL) and heated to reflux for 16 h. The reaction mixture was poured into ice-water and extracted with ethyl acetate (3 × 50 mL). The combined extracts were washed with water, saturated aqueous sodium bicarbonate, and brine, dried over anhydrous sodium sulfate, filtered, and evaporated. Purification by flash column chromatography on silica gel with DCM gave ester 2 (5.33 g, 42% over two steps) as a white solid: the obtained 1H, 12C and spectra matched to those reported by Tseng et al.35
5-(5-Bromothiophen-2-yl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxylic acid ethyl ester (3). To a magnetically stirred solution of 2 (2.60 g, 6.61 mmol) in acetonitrile (23 mL) was added NBS (1.43 g, 7.94 mmol) in small portions under nitrogen at 0 °C. The resulting mixture was then warmed to room temperature and stirred for 16 h. The reaction was quenched with saturated aqueous sodium thiosulfate and concentrated under reduced pressure to remove acetonitrile. The aqueous layer was extracted with ethyl acetate (2 × 40 mL). The organic layers were combined, washed with water, brine, dried over anhydrous sodium sulfate, and concentrated to give the crude residue, which was subjected to purification by flash chromatography on silica gel with n-hexane–ethyl acetate (8
:2) to afford bromo ester 3 (2.70 g, 90%) as a white solid: 1H NMR (400 MHz, chloroform-d) δ 7.46 (d, J = 2.0 Hz, 1H), 7.36 (d, J = 8.5 Hz, 1H), 7.33 (dd, J = 8.4, 2.0 Hz, 1H), 6.95 (dd, J = 3.9, 0.8 Hz, 1H), 6.64 (dd, J = 3.9, 0.8 Hz, 1H), 4.44 (q, J = 7.1 Hz, 2H), 2.42 (s, 3H), 1.42 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 162.5, 142.9, 136.9, 136.6, 135.7, 133.8, 130.9, 130.2, 130.1, 130.1, 129.3, 127.9, 120.4, 115.0, 61.1, 14.5, 10.0. ESMS, calculated m/z C17H13BrCl2N2O2S 459.92 [M]+, found m/z (relative intensity) 460.9 [M + H]+ (100), 482.9 [M + Na]+ (100).
5-(5-Bromothiophen-2-yl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxylic acid (4). To a solution of bromo ester 3 (2.64 g, 5.62 mmol) in methanol (26 mL) was added potassium hydroxide (0.73 g, 11.24 mmol) in methanol (8 mL) dropwise at room temperature. The resulting mixture was heated to reflux for 3 h. After hydrolysis was complete, the reaction mixture was cooled to room temperature, poured into ice-water, and acidified with 2N hydrochloric acid. The precipitate was filtered, washed with water, and dried under vacuum to give the thiophene carboxylic acid 4 (2.19 g, 90%) as a white solid: 1H NMR (400 MHz, DMSO-d6) δ 13.05 (s, 1H), 7.89 (d, J = 2.3 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.63 (dd, J = 8.5, 2.3 Hz, 1H), 7.23 (d, J = 3.9 Hz, 1H), 6.88 (d, J = 3.9 Hz, 1H), 2.32 (s, 3H); 13C NMR (101 MHz, DMSO) δ 163.9, 143.3, 136.7, 136.2, 135.9, 133.1, 132.3, 131.5, 130.8, 130.2, 130.0, 129.0, 119.6, 114.6, 10.2. ESMS, calculated m/z C15H9BrCl2N2O2S 431.9 [M]+, found m/z (relative intensity) 470.9 [M + K]+ (100).
General procedure for the synthesis of compounds 5a–5f. The general procedure is illustrated below for compound 5a.
5-(5-Bromothiophen-2-yl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride. A solution of the crude acid 4 (0.80 g, 1.81 mmol) and thionyl chloride (399 μL, 5.44 mmol) in toluene (12 mL) was refluxed for 3 h. Solvent was evaporated under reduced pressure, and the residue was then re-dissolved in toluene (8 mL) first and then in hexane (5 mL); after evaporation the crude acyl chloride (0.80 g, 98% yield) was obtained as a white solid.
5-(5-Bromothiophen-2-yl)-1-(2,4-dichlorophenyl)-4-methyl-N-(3-(trifluoromethyl)phenyl)-1H-pyrazole-3-carboxamide (5a). A solution in dichloromethane of the carboxylic chloride (1 mL, 0.36 M) obtained previously from 4, was added dropwise to a solution of 3-(trifluomethyl)aniline (50 μL, 0.40 mmol) and triethylamine (50 μL, 0.36 mmol) in dichloromethane (0.5 mL) at 0 °C. After stirring at room temperature for 16 h, to the reaction mixture was added water and the organic phase was extracted with dichloromethane (3 × 3 mL). The combined extracts were washed with brine, dried over anhydrous sodium sulfate, filtered, and evaporated. Flash column chromatography on silica gel with n-hexane–ethyl acetate (8
:2) gave carboxamide 5a (150 mg, 66% yield) as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.87 (s, 1H), 8.01 (s, 1H), 7.85 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.46 (t, J = 8.0 Hz, 1H), 7.39 (dd, J = 8.5, 2.1 Hz, 1H), 7.36 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 3.9 Hz, 1H), 6.68 (d, J = 3.9 Hz, 1H), 2.52 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.4, 144.3, 138.3, 137.7, 136.8, 135.5, 133.7, 131.42 (q, J = 32.4 Hz), 130.6, 130.4, 130.2, 129.9, 129.5, 129.4, 128.1, 125.2, 123.87 (q, J = 272.6 Hz), 122.57, 120.55 (q, J = 3.9 Hz), 119.6, 116.34 (q, J = 4.0 Hz), 115.2; 19F NMR (376 MHz, chloroform-d) δ −62.72. ESMS, calculated m/z C22H13BrCl2F3N3OS 574.93 [M]+, found m/z (relative intensity) 575.9 [M + H]+ (100). HRMS m/z M+H+ calcd for C22H13BrCl2F3N3OS: 573.9370; found: 573.9361%.
5-(5-Bromothiophen-2-yl)-1-(2,4-dichlorophenyl)-4-methyl-N-(3-(pentafluoro-λ6-sulfanyl)phenyl)-1H-pyrazole-3-carboxamide (5b). 5b (163 mg, 68% yield) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.87 (s, 1H), 8.13 (t, J = 2.0 Hz, 1H), 7.87 (dd, J = 8.1, 1.9 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 7.50 (d, J = 8.3 Hz, 1H), 7.44 (d, J = 8.1 Hz, 1H), 7.40 (dd, J = 8.5, 2.1 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 3.9 Hz, 1H), 6.68 (d, J = 3.9 Hz, 1H), 2.52 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.4, 154.25 (appt, J = 17.1 Hz), 144.2, 138.2, 137.8, 136.9, 135.4, 133.8, 130.6, 130.4, 130.3, 129.8, 129.4, 129.2, 128.1, 122.5, 121.28 (p, J = 4.7 Hz), 119.6, 117.25 (p, J = 4.0 Hz), 115.2, 9.7; 19F NMR (377 MHz, chloroform-d) δ 84.17 (p, J = 149.2 Hz), 62.73 (d, J = 150.0 Hz). ESMS, calculated m/z C21H13BrCl2F5N3OS2 632.90 [M]+, found m/z (relative intensity) 631.9 [M − H]− (100). HRMS m/z M+H+ calcd for C21H13BrCl2F5N3OS2: 631.9059; found: 631.9052%.
5-(5-Bromothiophen-2-yl)-N-(3-(tert-butyl)phenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (5c). 5c (87 mg, 63% yield) was obtained as a white solid 1H NMR (400 MHz, chloroform-d) δ 8.71 (s, 1H), 7.64–7.57 (m, 1H), 7.59 (d, J = 2.0 Hz, 1H), 7.54 (dd, J = 1.9, 0.7 Hz, 1H), 7.38 (dd, J = 1.9, 1.3 Hz, 2H), 7.30 (d, J = 8.6 Hz, 1H), 7.18–7.12 (m, 1H), 6.97 (d, J = 3.9 Hz, 1H), 6.67 (d, J = 3.8 Hz, 1H), 2.52 (s, 3H), 1.33 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 160.2, 152.2, 144.9, 137.5, 137.4, 136.7, 135.6, 133.8, 130.7, 130.4, 130.2, 130.1, 129.2, 128.6, 128.0, 121.2, 119.4, 117.0, 116.9, 115.0, 34.8, 31.3 (x 3), 9.8. ESMS, calculated m/z C25H22BrCl2N3OS 563.00 [M]+, found m/z (relative intensity) 564.0 [M + H]+ (100). HRMS m/z M+H+ calcd for C25H22BrCl2N3OS: 562.0122; found: 562.0111%.
5-(5-Bromothiophen-2-yl)-1-(2,4-dichlorophenyl)-4-methyl-N-(4-(trifluoromethyl)phenyl)-1H-pyrazole-3-carboxamide (5d). 5d (97 mg, 44% yield) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.89 (s, 1H), 7.80 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H), 7.54 (d, J = 2.1 Hz, 1H), 7.40 (dd, J = 8.5, 2.1 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 3.9 Hz, 1H), 6.68 (d, J = 3.9 Hz, 1H), 2.52 (d, J = 0.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 160.4, 144.3, 140.9, 137.7, 136.9, 135.4, 133.7, 130.6, 130.4, 130.2, 129.8, 129.4, 128.2, 126.25 (q, J = 3.6 Hz), 125.57 (q, J = 32.5 Hz, ×2), 124.13 (q, J = 272.2), 119.6, 119.2 (×2), 115.2, 9.7; 19F NMR (376 MHz, chloroform-d) δ −62.07. ESMS, calculated m/z C22H13BrCl2F3N3OS 574.93 [M]+, found m/z (relative intensity) 575.8 [M + H]+ (100). HRMS m/z M+H+ calcd for C22H13BrCl2F3N3OS: 573.9370; found: 573.9361%.
5-(5-Bromothiophen-2-yl)-1-(2,4-dichlorophenyl)-4-methyl-N-(4-(pentafluoro-λ6-sulfanyl)phenyl)-1H-pyrazole-3-carboxamide (5e). 5e (100 mg, 42% yield) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.92 (s, 1H), 7.81–7.68 (m, 4H), 7.53 (d, J = 2.1 Hz, 1H), 7.42–7.31 (m, 2H), 6.98 (d, J = 3.9 Hz, 1H), 6.68 (d, J = 3.9 Hz, 1H), 2.51 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.4, 149.03 (appt, J = 17.6 Hz), 144.1, 140.5, 137.8, 136.9, 135.4, 133.7, 130.6, 130.4, 130.2, 129.7, 129.4, 128.1, 126.99 (p, J = 4.7 Hz, ×2), 119.7, 118.8 (×2), 115.2, 9.7; 19F NMR (377 MHz, chloroform-d) δ 85.35 (p, J = 150.1, 149.4 Hz), 63.55 (d, J = 150.0 Hz). ESMS, calculated m/z C21H13BrCl2F5N3OS2 632.90 [M]+, found m/z (relative intensity) 631.8 [M − H]−. HRMS m/z M+H+ calcd for C21H13BrCl2F5N3OS2: 631.9059; found: 631.9048%.
5-(5-Bromothiophen-2-yl)-N-(4-(tert-butyl)phenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (5f). 5f (84 mg, 40% yield) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.67 (s, 1H), 7.58 (d, J = 8.7 Hz, 2H), 7.53 (dd, J = 1.9, 0.6 Hz, 1H), 7.41–7.32 (m, 4H), 6.97 (d, J = 3.9 Hz, 1H), 6.67 (d, J = 3.8 Hz, 1H), 2.52 (s, 3H), 1.32 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 160.2, 147.0, 144.9, 137.4, 136.7, 135.6, 135.2 (×2), 133.8, 130.7, 130.4, 130.2, 129.2, 128.0, 125.8 (×2), 119.6 (×2), 119.4, 115.0, 34.4, 31.4 (×3) 9.8. ESMS, calculated m/z C25H22BrCl2N3OS 563.0 [M]+, found m/z (relative intensity) 564.0 [M + H]+ (100). HRMS m/z M+H+ calcd for C25H22BrCl2N3OS: 562.0122; found: 562.0113%.
Lithium salt of ethyl 4-(5-bromothiophen-2-yl)-2,4-dioxobutanoate (6). To a magnetically stirred solution of lithium bis(tri-methylsilyl)amide (23 mL, 22.56 mmol, 1.0 M in THF) in diethyl ether (41 mL) at −78 °C, 1-(5-bromothiophen-2-yl)ethanone (4.25 g, 20.51 mmol) in diethyl ether (16 mL) was added dropwise under a nitrogen atmosphere. After the mixture was stirred at the same temperature for an additional 45 min, diethyl oxalate (3.4 mL, 24.62 mmol) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for another 16 h. The reaction precipitate was filtered, washed with diethyl ether, and dried under vacuum to afford the crude lithium salt 6 (6.31 g, 99%) as a pale-yellow solid. The product was used without further purification. ESMS, calculated m/z C10H8BrLiO4S 309.9 [M]+, found m/z (relative intensity) 326.9 [M − Li + H + Na]+ (63), 343 [M − Li + H + K]+ (31).
General procedure for the synthesis of compounds 7a,7b. The general procedure is illustrated below for compound 7a.
Ethyl 5-(5-bromothiophen-2-yl)-1-(2,4-difluorophenyl)-1H-pyrazole-3-carboxylate (7a). To a solution of lithium salt 6 (3 g, 9.55 mmol) in ethanol (26 mL) was added 2,4-difluorophenylhydrazine hydrochloride (2.143 g, 11.46 mmol) in one portion at room temperature under nitrogen. The resulting mixture was stirred at the same temperature for 24 h. After reaction was complete, the precipitate was filtered, washed with ethanol and diethyl ether, and dried under vacuum to give a light-yellow solid (2.66 g). This crude solid, without purification, was dissolved in acetic acid (20 mL) and heated to reflux for 16 h. The reaction mixture was poured into ice-water and extracted with ethyl acetate (3 × 70 mL). The combined extracts were washed with water, saturated aqueous sodium bicarbonate, and brine, dried over anhydrous sodium sulfate, filtered, and evaporated. Purification by flash column chromatography on silica gel with DCM gave ester 7a (2.08 g, 58% over two steps) as a white solid. 1H NMR (400 MHz, chloroform-d) δ 7.51 (td, J = 8.5, 5.7 Hz, 1H), 7.08 (s, 1H), 7.08–6.92 (m, 2H), 6.93 (d, J = 3.9 Hz, 1H), 6.69 (d, J = 3.9 Hz, 1H), 4.44 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 163.54 (dd, J = 254.1, 11.0 Hz), 161.6, 157.88 (dd, J = 256.9, 12.8 Hz), 145.3, 139.5, 130.83 (d, J = 10.1 Hz), 130.8, 130.4, 127.4, 123.29 (dd, J = 12.7, 4.1 Hz), 114.4, 112.25 (dd, J = 22.7, 3.8 Hz), 108.4, 105.29 (dd, J = 26.5, 23.0 Hz), 61.3, 14.3; 19F NMR (376 MHz, chloroform-d) δ −104.59 (qd, J = 8.4, 5.7 Hz), −115.10 to −115.23 (m). ESMS, calculated m/z C16H11BrF2N2O2S 414.0 [M]+, found m/z (relative intensity) 415.0 [M + H]+ (100), 435.0 [M + Na]+ (100).
Ethyl 5-(5-bromothiophen-2-yl)-1-(2,4-dichlorophenyl)-1H-pyrazole-3-carboxylate (7b). 7b (2.36 g, 60% over two steps) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 7.54 (d, J = 2.1 Hz, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.41 (dd, J = 8.5, 2.1 Hz, 1H), 7.08 (s, 1H), 6.92 (d, J = 3.9 Hz, 1H), 6.66 (d, J = 3.9 Hz, 1H), 4.44 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 161.7, 145.2, 139.5, 137.2, 135.3, 134.0, 131.0, 130.8, 130.5, 130.4, 128.2, 127.3, 114.5, 108.1, 61.4, 14.4. ESMS, calculated m/z 16H11BrCl2N2O2S 445.9 [M]+, found m/z (relative intensity) 446.9 [M + H]+ (100).
General procedure for the synthesis of compounds 8a,8b. The general procedure is illustrated below for compound 8a.
5-(5-Bromothiophen-2-yl)-1-(2,4-difluorophenyl)-1H-pyrazole-3-carboxylic acid (8a). To a solution of bromo ester 7a (1.72 g, 4.07 mmol) in methanol (19 mL) was added potassium hydroxide (0.80 g, 12.21 mmol) in methanol (9 mL) dropwise at room temperature. The resulting mixture was heated to reflux for 3 h. After hydrolysis was complete, the reaction mixture was cooled to room temperature, poured into ice-water, and acidified with 2N hydrochloric acid. The precipitate was filtered, washed with water, and dried under vacuum to give thiophene carboxylic acid 8a (1.34 g, 85%) as a white solid: 1H NMR (400 MHz, DMSO-d6) δ 13.18 (bs, 1H), 7.83 (td, J = 8.8, 5.9 Hz, 1H), 7.64 (ddd, J = 10.3, 9.0, 2.8 Hz, 1H), 7.42–7.32 (m, 1H), 7.28 (s, 1H), 7.23 (d, J = 4.0 Hz, 1H), 7.11 (d, J = 4.0 Hz, 1H); 13C NMR (101 MHz, DMSO) δ 163.66 (dd, J = 251.4, 11.7 Hz), 163.0, 157.92 (dd, J = 253.6, 13.7 Hz), 146.0, 139.7, 132.28 (d, J = 10.7 Hz), 131.6, 130.8, 129.3, 123.43 (dd, J = 12.6, 3.8 Hz), 114.0, 113.45 (dd, J = 22.9, 3.6 Hz), 108.6, 106.16 (dd, J = 27.4, 23.6 Hz); 19F NMR (376 MHz, DMSO-d6) δ −104.80 (qd, J = 8.7, 5.8 Hz), −117.33 (q, J = 9.4 Hz). ESMS, calculated m/z C14H7BrF2N2O2S 383.9 [M]+, found m/z (relative intensity) 422.9 [M + K]+ (100).
5-(5-Bromothiophen-2-yl)-1-(2,4-dichlorophenyl)-1H-pyrazole-3-carboxylic acid (8b). 8b (1.42 g, 79%) was obtained as a white solid: 1H NMR (400 MHz, DMSO-d6) δ 13.18 (s, 1H), 8.00 (d, J = 2.3 Hz, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.71 (dd, J = 8.5, 2.3 Hz, 1H), 7.31 (s, 1H), 7.22 (d, J = 4.0 Hz, 1H), 7.09 (d, J = 4.0 Hz, 1H); 13C NMR (101 MHz, DMSO) δ 163.0, 145.8, 139.5, 136.9, 135.5, 133.6, 132.5, 131.6, 130.8, 130.6, 129.4, 129.1, 114.0, 108.3. ESMS, calculated m/z C14H7BrCl2N2O2S 417.9 [M]+, found m/z (relative intensity) 456.9 [M + K]+ (100).
General procedure for the synthesis of compounds 9a–9d. The general procedure is illustrated below for compound 9a.
5-(5-Bromothiophen-2-yl)-1-(2,4-difluorophenyl)-1H-pyrazole-3-carbonyl chloride. A solution of the crude acid 8 (0.20 g, 0.51 mmol) and thionyl chloride (112 μL, 1.53 mmol) in toluene (3 mL) was refluxed for 3 h. The solvent was evaporated under reduced pressure, and the residue was then re-dissolved in toluene (3 mL) first and then in hexane (4 mL); after evaporation the crude acyl chloride (0.19 g, 95% yield) was obtained as a white solid.
5-(5-Bromothiophen-2-yl)-1-(2,4-difluorophenyl)-N-(4-(trifluoromethyl)phenyl)-1H-pyrazole-3-carboxamide (9a). A solution in dichloromethane of the acyl chloride obtained previously (0.9 mL, 0.24 M), was added dropwise to a solution of 4-(trifluomethyl)aniline (30 μL, 0.23 mmol) and triethylamine (33 μL, 0.23 mmol) in dichloromethane (0.2 mL) at 0 °C. After stirring at room temperature for 16 h, to the reaction mixture was added water and the organic phase was extracted with dichloromethane (3 × 3 mL). The combined extracts were washed with brine, dried over anhydrous sodium sulfate, filtered, and evaporated. Flash column chromatography on silica gel with n-hexane–ethyl acetate (9
:1) gave carboxamide 9a (88 mg, 72% yield) as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.81 (s, 1H), 7.81 (d, J = 8.5 Hz, 2H), 7.61 (d, J = 8.6 Hz, 2H), 7.51 (td, J = 8.5, 5.7 Hz, 1H), 7.17 (s, 1H), 7.12–7.00 (m, 2H), 6.95 (d, J = 3.9 Hz, 1H), 6.74 (d, J = 3.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 163.74 (dd, J = 254.8, 10.9 Hz), 159.1, 158.03 (dd, J = 257.3, 12.8 Hz), 147.8, 140.6, 130.7, 130.64 (d, J = 10.2 Hz), 130.5, 127.7, 126.32 (q, J = 3.8 Hz, ×2), 125.99 (q, J = 32.6 Hz), 124.09 (q, J = 271.8 Hz), 123.15 (dd, J = 13.0, 4.2 Hz), 122.74, 119.3 (×2), 114.8, 112.48 (dd, J = 22.8, 3.9 Hz), 107.0, 105.66 (dd, J = 26.5, 23.0 Hz); 19F NMR (376 MHz, chloroform-d) δ −62.11, −104.02 (qd, J = 8.3, 5.7 Hz), −115.07 to −115.16 (m). ESMS, calculated m/z C21H11BrF5N3OS 529 [M]+, found m/z (relative intensity) 530 [M + H]+ (100). HRMS m/z M+H+ calcd for C21H11BrF5N3OS: 527.9805; found: 527.9794%.
5-(5-Bromothiophen-2-yl)-1-(2,4-dichlorophenyl)-N-(4-(trifluoromethyl)phenyl)-1H-pyrazole-3-carboxamide (9b). 9b (69 mg, 53%) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.81 (s, 1H), 7.80 (d, J = 8.5 Hz, 2H), 7.65–7.56 (m, 3H), 7.46 (d, J = 1.9 Hz, 2H), 7.18 (s, 1H), 6.94 (d, J = 3.9 Hz, 1H), 6.71 (d, J = 3.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 159.1, 147.7, 140.6, 140.5, 137.5, 135.1, 134.1, 130.7, 130.7, 130.7, 130.5, 128.4, 127.6, 126.33 (q, J = 3.7 Hz, ×2), 125.98 (q, J = 32.8 Hz), 124.08 (q, J = 271.3 Hz), 119.3 (×2), 114.8, 106.6; 19F NMR (376 MHz, chloroform-d) δ −62.10. ESMS, calculated m/z C21H11BrCl2F3N3OS 560.9 [M]+, found m/z (relative intensity) 583.8 [M + Na]+ (100). HRMS m/z M+H+ calcd for C21H11BrCl2F3N3OS: 559.9214; found: 559.9203%.
5-(5-Bromothiophen-2-yl)-1-(2,4-difluorophenyl)-N-(4-(pentafluoro-λ6-sulfanyl)phenyl)-1H-pyrazole-3-carboxamide (9c). 9c (110 mg, 70%) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.81 (s, 1H), 7.81–7.72 (m, 4H), 7.51 (td, J = 8.5, 5.7 Hz, 1H), 7.18 (s, 1H), 7.13–7.01 (m, 2H), 6.96 (d, J = 3.9 Hz, 1H), 6.74 (d, J = 3.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 163.76 (dd, J = 254.8, 11.0 Hz), 159.1, 158.02 (dd, J = 257.2, 12.8 Hz), 149.14 (appt, J = 17.5 Hz), 147.6, 140.7, 140.3, 130.63 (d, J = 10.4 Hz), 130.5, 127.7, 127.04 (p, J = 4.6 Hz, ×2), 123.11 (dd, J = 12.6, 4.2 Hz), 118.9 (×2), 114.9, 112.50 (dd, J = 22.8, 3.9 Hz), 107.0, 105.67 (dd, J = 26.4, 22.9 Hz), 100; 19F NMR (377 MHz, chloroform-d) δ 85.18 (p, J = 150.6, 149.7 Hz), 63.49 (d, J = 150.0 Hz), −103.91 (qd, J = 8.3, 5.7 Hz), −115.12 (q, J = 8.9 Hz). ESMS, calculated m/z C20H11BrF7N3OS2 586.9 [M]+, found m/z (relative intensity) 585.9 [M − H]− (100). HRMS m/z M+H+ calcd for C20H11BrF7N3OS2: 585.9493; found: 585.9484%.
5-(5-Bromothiophen-2-yl)-1-(2,4-dichlorophenyl)-N-(4-(pentafluoro-λ6-sulfanyl)phenyl)-1H-pyrazole-3-carboxamide (9d). 9d (60 mg, 40%) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.82 (s, 1H), 7.82–7.70 (m, 4H), 7.61 (d, J = 1.9 Hz, 1H), 7.52–7.40 (m, 2H), 7.19 (s, 1H), 6.95 (d, J = 3.9 Hz, 1H), 6.71 (d, J = 3.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 159.1, 149.13 (p, J = 17.9 Hz), 147.5, 140.6, 140.2, 137.6, 135.1, 134.1, 130.7, 130.7, 130.6, 130.5, 128.4, 127.6, 127.07 (p, J = 4.3 Hz, ×2), 118.9 (×2), 114.9, 106.6; 19F NMR (377 MHz, chloroform-d) δ 85.17 (p, J = 150.7, 149.7 Hz), 63.48 (d, J = 150.0 Hz). ESMS, calculated m/z C20H11BrCl2F5N3OS2 618.9 [M]+, found m/z (relative intensity) 617.8 [M − H]− (100). HRMS m/z M+H+ calcd for C20H11BrCl2F5N3OS2: 617.8902; found: 617.8894%.
Lithium salt of ethyl 4-(4-chlorophenyl)-3-methyl-2,4-dioxobutanoate (10). To a magnetically stirred solution of lithium bis(tri-methylsilyl)amide (64 mL, 63.93 mmol, 1.0 M in THF) in diethyl ether (63 mL) at −78 °C, 1-(2-thienyl)-1-propanone (10 g, 58.12 mmol) in diethyl ether (73 mL) was added dropwise under a nitrogen atmosphere. After the mixture was stirred at the same temperature for an additional 45 min, diethyl oxalate (9 mL, 63.93 mmol) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for another 16 h. The reaction precipitate was filtered, washed with diethyl ether, and dried under vacuum to afford the crude lithium salt 10 (8.05 g, 50%) as a pale-yellow solid. The product was used without further purification.
Ethyl 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxylate (11). To a solution of lithium salt 10 (8.05 g, 28.73 mmol) in ethanol (99 mL) was added 2,4- dichlorophenylhydrazine hydrochloride (6.88 g, 31.60 mmol) in one portion at room temperature under nitrogen. The resulting mixture was stirred at the same temperature for 24 h. After reaction was complete, the precipitate was filtered, washed with ethanol and diethyl ether, and dried under vacuum to give a light-yellow solid (6.1 g). This crude solid, without purification, was dissolved in acetic acid (45 mL) and heated to reflux for 16 h. The reaction mixture was poured into ice-water and extracted with ethyl acetate (3 × 50 mL). The combined extracts were washed with water, saturated aqueous sodium bicarbonate, and brine, dried over anhydrous sodium sulfate, filtered, and evaporated. Purification by flash column chromatography on silica gel with hexane–acetate (8
:2) gave ester 11 (3.44 g, 30% over two steps) as a white solid: 1H NMR (400 MHz, chloroform-d) δ 7.38 (d, J = 2.2 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.32–7.27 (m, 3H), 7.07 (d, J = 8.6 Hz, 2H), 4.45 (q, J = 7.1 Hz, 2H), 2.33 (s, 3H), 1.43 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 163.2, 143.4, 143.3, 136.5, 136.3, 135.4, 133.5, 131.3 (×2), 131.2, 130.5, 129.3 (×2), 128.2, 127.5, 119.6, 61.4, 14.9, 10.1. ESMS, calculated m/z C19H15Cl3N2O2 408.0 [M]+, found m/z (relative intensity) 409.0 [M + H]+ (100).
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxylic acid (12). To a solution of bromo ester 3 (2.32 g, 5.54 mmol) in methanol (26 mL) was added potassium hydroxide (0.72 g, 11.09 mmol) in methanol (8 mL) dropwise at room temperature. The resulting mixture was heated to reflux for 3 h. After hydrolysis was complete, the reaction mixture was cooled to room temperature, poured into ice-water, and acidified with 2N hydrochloric acid. The precipitate was filtered, washed with water, and dried under vacuum to give the carboxylic acid 12 (2.10 g, 98%) as a white solid: 1H NMR (400 MHz, DMSO-d6) δ 7.73 (d, J = 2.3 Hz, 1H), 7.67 (d, J = 8.5 Hz, 1H), 7.54 (dd, J = 8.5, 2.3 Hz, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.21 (d, J = 8.4 Hz, 2H), 2.20 (s, 3H); 13C NMR (101 MHz, DMSO) δ 164.1, 143.4, 142.9, 136.1, 135.6, 134.3, 132.3, 132.1, 131.8 (×2), 130.0, 129.2 (×2), 128.8, 127.4, 118.4, 9.9. ESMS, calculated m/z C17H11Cl3N2O2 380.0 [M]+, found m/z (relative intensity) 381.0 [M + H]+ (100).
General procedure for the synthesis of compounds 13a–13g. The general procedure is illustrated below for compound 13a.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride. A solution of the crude acid 12 (1 g, 2.57 mmol) and thionyl chloride (565 μL, 7.70 mmol) in toluene (17.1 mL) was refluxed for 3 h. Solvent was evaporated under reduced pressure, and the residue was then re-dissolved in toluene (10 mL) first and then in hexane (10 mL); after evaporation the crude acyl chloride (0.98 g, 95% yield) was obtained as a white solid.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(3-(trifluoromethyl)phenyl)-1H-pyrazole-3-carboxamide (13a). A solution in dichloromethane of the acyl chloride obtained previously (0.96 mL, 0.41 M), was added dropwise to a solution of 3-(trifluomethyl)aniline (55 μL, 0.44 mmol) and triethylamine (61 μL, 0.44 mmol) in dichloromethane (0.4 mL) at 0 °C. After stirring at room temperature for 16 h, to the reaction mixture was added water and the organic phase was extracted with dichloromethane (3 × 3 mL). The combined extracts were washed with brine, dried over anhydrous sodium sulfate, filtered, and evaporated. Flash column chromatography on silica gel with n-hexane–ethyl acetate (8
:2) gave carboxamide 13a (160 mg, 70% yield) as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.91 (s, 1H), 8.03 (t, J = 1.9 Hz, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.51–7.41 (m, 2H), 7.40–7.27 (m, 5H), 7.12–7.07 (d, J = 8.5 Hz, 2H), 2.43 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.6, 144.4, 143.7, 138.4, 136.3, 135.7, 135.2, 133.0, 131.40 (q, J = 32.3 Hz), 130.8 (×2), 130.5, 130.4, 129.5, 129.0 (×2), 128.0, 126.8, 122.55, 122.53 (q, J = 272.0 Hz), 120.50 (q, J = 3.9 Hz), 118.4, 116.32 (q, J = 4.0 Hz), 9.5; 19F NMR (376 MHz, chloroform-d) δ −62.74. ESMS, calculated m/z C24H15Cl3F3N3O 523 [M]+, found m/z (relative intensity) 546.0 [M + Na]+ (100). HRMS m/z M+H+ calcd for C24H15Cl3F3N3O: 524.0311; found: 524.0301%.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(3-(pentafluoro-λ6-sulfanyl)phenyl)-1H-pyrazole-3-carboxamide (13b). 13b (182 mg, 70% yield) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.92 (s, 1H), 8.16 (t, J = 2.1 Hz, 1H), 7.91 (d, J = 9.0 Hz, 1H), 7.55–7.43 (m, 3H), 7.39–7.31 (m, 4H), 7.12 (d, J = 8.5 Hz, 2H), 2.45 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.6, 154.25 (appt, J = 17.4 Hz), 144.2, 143.7, 138.3, 136.3, 135.7, 135.2, 133.0, 130.8 (×2), 130.5, 130.4, 129.2, 129.0 (×2), 128.0, 126.8, 122.4, 121.24 (p, J = 3.9, 3.4 Hz), 118.4, 117.22 (p, J = 4.9 Hz), 9.5; 19F NMR (377 MHz, chloroform-d) δ 84.17 (p, J = 150.4 Hz), 62.72 (d, J = 150.0 Hz). ESMS, calculated m/z C23H15Cl3F5N3OS 583.0 [M]+, found m/z (relative intensity) 582.0 [M − H]− (56). HRMS m/z M+H+ calcd for C23H15Cl3F5N3OS: 582.0000; found: 581.9991%.
N-(3-(tert-Butyl)phenyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (13c). 13c (88 mg, 63% yield) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.76 (s, 1H), 7.64–7.59 (m, 2H), 7.46 (t, J = 1.3 Hz, 1H), 7.35–7.25 (m, 5H), 7.18–7.13 (m, 1H), 7.10 (d, J = 8.5 Hz, 2H), 2.44 (s, 3H), 1.33 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 160.4, 152.2, 145.0, 143.4, 137.6, 136.2, 135.8, 135.0, 133.1, 130.9 (×2), 130.6, 130.4, 128.9 (×2), 128.6, 128.0, 127.1, 121.1, 118.2, 117.1, 116.9, 34.8, 31.4 (×3), 9.6. ESMS, calculated m/z C27H24Cl3N3O 511.1 [M]+, found m/z (relative intensity) 512.1 [M + H]+ (100). HRMS m/z M+H+ calcd for C27H24Cl3N3O: 512.1063; found: 512.1052%.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(4-(trifluoromethyl)phenyl)-1H-pyrazole-3-carboxamide (13d). 13d (141 mg, 58% yield) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.93 (s, 1H), 7.82 (d, J = 8.3 Hz, 2H), 7.60 (d, J = 8.3 Hz, 2H), 7.47 (s, 1H), 7.37–7.28 (m, 4H), 7.09 (d, J = 8.5 Hz, 2H), 2.43 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.6, 144.4, 143.7, 141.0, 136.3, 135.7, 135.2, 133.0, 130.8 (×2), 130.5, 129.0 (×2), 128.2, 128.0, 126.8, 126.27 (q, J = 3.7 Hz, ×2), 125.55 (q, J = 31.9 Hz), 124.93 (q, J = 272.0 Hz), 119.2 (×2), 118.5, 9.5. ESMS, calculated m/z C24H15Cl3F3N3O 523 [M]+, found m/z (relative intensity) 524.0 [M + H]+ (100). HRMS m/z M+H+ calcd for C24H15Cl3F3N3O: 524.0311; found: 524.0300%.
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(4-(pentafluoro-λ6-sulfanyl)phenyl)-1H-pyrazole-3-carboxamide (13e). 13e (177 mg, 68% yield) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.93 (s, 1H), 7.79 (d, J = 9.0 Hz, 2H), 7.77–7.69 (m, 2H), 7.47 (d, J = 2.1 Hz, 1H), 7.37–7.28 (m, 4H), 7.09 (d, J = 8.4 Hz, 2H), 2.43 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.6, 148.97 (appt, J = 17.0 Hz), 144.2, 143.8, 140.6, 136.4, 135.6, 135.2, 133.0, 130.8 (×2), 130.43, 130.42, 129.0 (×2), 128.0, 126.97 (p, J = 4.6 Hz, ×2), 126.72, 118.8 (×2), 118.5, 9.5; 19F NMR (377 MHz, chloroform-d) δ 85.34 (p, J = 150.1, 149.1 Hz), 63.53 (d, J = 149.9 Hz). ESMS, calculated m/z C23H15Cl3F5N3OS 5823.0 [M]+, found m/z (relative intensity) 582.0 [M − H]− (64). HRMS m/z M+H+ calcd for C23H15Cl3F5N3OS: 582.0000; found: 581.9990%.
N-(4-(tert-Butyl)phenyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (13f). 13f (162 mg, 78% yield) was obtained as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.71 (s, 1H), 7.60 (d, J = 8.7 Hz, 2H), 7.46 (dd, J = 1.8, 0.8 Hz, 1H), 7.36 (d, J = 8.7 Hz, 2H), 7.33–7.29 (m, 4H), 7.09 (d, J = 8.5 Hz, 2H), 2.43 (s, 3H), 1.32 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 160.4, 147.0, 145.0, 143.4, 136.1, 135.9, 135.2, 135.0, 133.0, 130.9 (×2), 130.6, 130.4, 128.9 (×2), 127.9, 127.1, 125.8 (×2), 119.5 (×2), 118.2, 34.4, 31.4 (×3), 9.5. ESMS, calculated m/z C27H24Cl3N3O 511.1 [M]+, found m/z (relative intensity) 534.1 [M + Na]+ (100). HRMS m/z M+H+ calcd for C27H24Cl3N3O: 512.1063; found: 512.1053%.
Determination of the partition coefficients (log
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) P). Measurement of chromatographic capacity factor (k′) by C-18 HPLC method was performed using the equation k′ = (tr − t0)/t0,47,48 where t0 is the retention time of unretained substance and tr is the compound's retention time. The mobile phase was prepared mixing methanol with water in proportions of 85:15 and the flow rate was 1 mL min−1. A solution of urea in a methanol–water solvent (85:15) was used for measurement of the column dead time (t0 = 2.673 ± 0.002, n = 3). Seven compounds having known logP values, tabulated and in the range from 1.10 to 5.70 (benzyl alcohol, logP 1.10; benzene, logP 2.10; toluene, logP 2.70; naphthalene, logP 3.60; biphenyl, logP 4; phenanthrene, logP 4.50; triphenylamine, logP 5.70) were chosen as a “standard” calibration mixture for the determination of retention times (tr). Every measure was obtained in triplicate and at controlled temperature of 25 °C. Capacity factors (k′) were calculated. The log k' value for each of seven compounds was plotted against its relative lipophilicity value reported in literature, based on the established linear relationship (logP = 3.64 log k′ + 3.39, correlation coefficient = 0.95). The capacity factor of each compound was determined (the value was obtained on average of three experiments) and the relative logP value was obtained by extrapolation.
P). Measurement of chromatographic capacity factor (k′) by C-18 HPLC method was performed using the equation k′ = (tr − t0)/t0,47,48 where t0 is the retention time of unretained substance and tr is the compound's retention time. The mobile phase was prepared mixing methanol with water in proportions of 85:15 and the flow rate was 1 mL min−1. A solution of urea in a methanol–water solvent (85:15) was used for measurement of the column dead time (t0 = 2.673 ± 0.002, n = 3). Seven compounds having known logP values, tabulated and in the range from 1.10 to 5.70 (benzyl alcohol, logP 1.10; benzene, logP 2.10; toluene, logP 2.70; naphthalene, logP 3.60; biphenyl, logP 4; phenanthrene, logP 4.50; triphenylamine, logP 5.70) were chosen as a “standard” calibration mixture for the determination of retention times (tr). Every measure was obtained in triplicate and at controlled temperature of 25 °C. Capacity factors (k′) were calculated. The log k' value for each of seven compounds was plotted against its relative lipophilicity value reported in literature, based on the established linear relationship (logP = 3.64 log k′ + 3.39, correlation coefficient = 0.95). The capacity factor of each compound was determined (the value was obtained on average of three experiments) and the relative logP value was obtained by extrapolation.
In vitro assays
β-Arrestin assays
PathHunter® HEK293 CB1 beta-arrestin cells were plated 48 hours before use and incubated at 37 °C, 5% CO2 in a humidified incubator. Compounds were dissolved in DMSO and diluted in OCC media to the required concentrations. 5 μL of compound or vehicle solution was added to each well and incubated for 60 minutes at 37 °C, 5% CO2 in a humidified incubator. 5 μL of increasing concentrations of CP55940 was added to each well followed by a 90 minute incubation at 37 °C, 5% CO2 in a humidified incubator. 55 μL of detection reagent is then added followed by a further 90 minute incubation at room temperature in the dark. Chemiluminescence, indicated as RLU, was measured on a standard luminescence plate reader.Solubility tests
The aqueous solubility was measured using laser nephelometry (BMG Labtech Nephelometer) following serial dilution of DMSO stocks into Tris Buffer (50 mM Tris–HCL, 50 mM Tris base and 0.1% BSA) to give final concentrations of 0.01, 0.1,1 and 10 μM and a final DMSO concentration of 0.1%. The amount of laser scatter caused by insoluble particulates (relative nephelometry units) was measured. RFU values 3-fold greater than control (0 μM) indicate insolubility.Acknowledgements
We thank the European Commission for financial support (Industry Academia Partnerships and Pathways project “PET BRAIN”, Contract no. 251482) and Miteni S.p.A. (Italy) for a kind gift of the SF5 precursors. The EPSRC National Mass Spectrometry Service Centre (Swansea, UK) is gratefully acknowledged for performing HRMS analyses. We thank Dr Kevin Read (University of Dundee) for support and advice with the nephelometry assays. S.A. thanks the Northern Research Partnership (http://www.northscotland-research.ac.uk/) and Pharmaness (http://www.pharmaness.it) for co-funding a PhD studentship.Notes and references
- D. O'Hagan and D. B. Harper, J. Fluorine Chem., 1999, 100, 127–133 CrossRef CAS.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- M. Zanda, New J. Chem., 2004, 28, 1401–1411 RSC.
- D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071–1081 CrossRef CAS PubMed.
- W. A. Sheppard, J. Am. Chem. Soc., 1960, 82, 4751–4752 CrossRef CAS.
- J. T. Welch, in Fluorine In Pharmaceutical And Medicinal Chemistry, Imperial college press, 2012, vol. 6, pp. 175–207 Search PubMed.
- S. Altomonte and M. Zanda, J. Fluorine Chem., 2012, 143, 57–93 CrossRef CAS PubMed.
- R. W. Winter, R. A. Dodean, and G. L. Gard, in Fluorine-Containing Synthons, ed. V. A. Soloshonok, American Chemical Society, Washington, DC, 2005, vol. 911, pp. 87–118 Search PubMed.
- W. A. Sheppard, J. Am. Chem. Soc., 1965, 87, 2410–2420 CrossRef CAS.
- C. Hansch, R. M. Muir, T. Fujita, P. P. Maloney, F. Geiger and M. Streich, J. Am. Chem. Soc., 1963, 85, 2817–2824 CrossRef CAS.
- W. A. Sheppard, J. Am. Chem. Soc., 1962, 84, 3072–3076 CrossRef CAS.
- J. T. Welch and D. S. Lim, Bioorg. Med. Chem., 2007, 15, 6659–6666 CrossRef CAS PubMed.
- P. Kirsch, Modern fluoroorganic chemistry synthesis, reactivity, applications, Wiley-VCH, Weinheim, 2004 Search PubMed.
- D. Lentz and K. Seppelt, in Chemistry of Hypervalent Compounds, ed. K. Akiba, Wiley-VCH, New York, 1999, pp. 295–323 Search PubMed.
- L. J. Sæthre, N. Berrah, J. D. Bozek, K. J. Børve, T. X. Carroll, E. Kukk, G. L. Gard, R. Winter and T. D. Thomas, J. Am. Chem. Soc., 2001, 123, 10729–10737 CrossRef PubMed.
- A. Kapur, P. Samaniego, G. A. Thakur, A. Makriyannis and M. E. Abood, J. Pharmacol. Exp. Ther., 2008, 325, 341–348 CrossRef CAS PubMed.
- S. Munro, K. L. Thomas and M. Abu-Shaar, Nature, 1993, 365, 61–65 CrossRef CAS PubMed.
- A. C. Howlett, F. Barth, T. I. Bonner, G. Cabral, P. Casellas, W. A. Devane, C. C. Felder, M. Herkenham, K. Mackie, B. R. Martin, R. Mechoulam and R. G. Pertwee, Pharmacol. Rev., 2002, 54, 161–202 CrossRef CAS.
- D. Shire, C. Carillon, M. Kaghad, B. Calandra, M. Rinaldi-Carmona, G. L. Fur, D. Caput and P. Ferrara, J. Biol. Chem., 1995, 270, 3726–3731 CrossRef CAS PubMed.
- E. Ryberg, H. K. Vu, N. Larsson, T. Groblewski, S. Hjorth, T. Elebring, S. Sjögren and P. J. Greasley, FEBS Lett., 2005, 579, 259–264 CrossRef CAS PubMed.
- M. Herkenham, A. B. Lynn, M. D. Little, M. R. Johnson, L. S. Melvin, B. R. de Costa and K. C. Rice, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 1932–1936 CrossRef CAS.
- G. Griffin, S. R. Fernando, R. A. Ross, N. G. McKay, M. L. J. Ashford, D. Shire, J. W. Huffman, S. Yu, J. A. H. Lainton and R. G. Pertwee, Eur. J. Pharmacol., 1997, 339, 53–61 CrossRef CAS.
- R. G. Pertwee, Life Sci., 1999, 65, 597–605 CrossRef CAS.
- J. C. Ashton, D. Friberg, C. L. Darlington and P. F. Smith, Neurosci. Lett., 2006, 396, 113–116 CrossRef CAS PubMed.
- J. Horder, M. Browning, M. Di Simplicio, P. J. Cowen and C. J. Harmer, J. Psychopharmacol., 2012, 26, 125–132 CrossRef CAS PubMed.
- G. Kunos, D. Osei-Hyiaman, S. Bátkai, K. A. Sharkey and A. Makriyannis, Trends Pharmacol. Sci., 2009, 30, 1–7 CrossRef CAS PubMed.
- E. Kirilly, X. Gonda and G. Bagdy, Acta Physiol., 2012, 205, 41–60 CrossRef CAS.
- B.-C. Ho, T. H. Wassink, S. Ziebell and N. C. Andreasen, Schizophr. Res., 2011, 128, 66–75 CrossRef PubMed.
- B. Costa, A. E. Trovato, M. Colleoni, G. Giagnoni, E. Zarini and T. Croci, Pain, 2005, 116, 52–61 CrossRef CAS PubMed.
- P. Gazzerro, M. G. Caruso, M. Notarnicola, G. Misciagna, V. Guerra, C. Laezza and M. Bifulco, Int. J. Obes., 2006, 31, 908–912 CrossRef PubMed.
- M. Rinaldi-Carmona, F. Barth, M. Héaulme, D. Shire, B. Calandra, C. Congy, S. Martinez, J. Maruani, G. Néliat, D. Caput, P. Ferrara, P. Soubrié, J. C. Brelière and G. Le Fur, FEBS Lett., 1994, 350, 240–244 CrossRef CAS.
- R. Lan, Q. Liu, P. Fan, S. Lin, S. R. Fernando, D. McCallion, R. Pertwee and A. Makriyannis, J. Med. Chem., 1999, 42, 769–776 CrossRef CAS PubMed.
- P. Lazzari, S. Ruiu, G. A. Pinna and G. Murineddu, US Pat., 0 261 281, 2005 Search PubMed.
- R. D. Bowden, P. J. Comina, M. P. Greenhall, B. M. Kariuki, A. Loveday and D. Philp, Tetrahedron, 2000, 56, 3399–3408 CrossRef CAS.
- S.-L. Tseng, M.-S. Hung, C.-P. Chang, J.-S. Song, C.-L. Tai, H.-H. Chiu, W.-P. Hsieh, Y. Lin, W.-L. Chung, C.-W. Kuo, C.-H. Wu, C.-M. Chu, Y.-S. Tung, Y.-S. Chao and K.-S. Shia, J. Med. Chem., 2008, 51, 5397–5412 CrossRef CAS PubMed.
- A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525–616 CrossRef CAS.
- H. Fan, H. T. Ravert, D. P. Holt, R. F. Dannals and A. G. Horti, J. Labelled Compd. Radiopharm., 2006, 49, 1021–1036 CrossRef CAS.
- S. Tobiishi, T. Sasada, Y. Nojiri, F. Yamamoto, T. Mukai, K. Ishiwata and M. Maeda, Chem. Pharm. Bull., 2007, 55, 1213–1217 CrossRef CAS.
- R. A. Ross, H. C. Brockie, L. A. Stevenson, V. L. Murphy, F. Templeton, A. Makriyannis and R. G. Pertwee, Br. J. Pharmacol., 1999, 126, 665–672 CrossRef CAS PubMed.
- J.-Z. Chen, X.-W. Han, Q. Liu, A. Makriyannis, J. Wang and X.-Q. Xie, J. Med. Chem., 2006, 49, 625–636 CrossRef CAS PubMed.
- M. R. Price, G. L. Baillie, A. Thomas, L. A. Stevenson, M. Easson, R. Goodwin, A. McLean, L. McIntosh, G. Goodwin, G. Walker, P. Westwood, J. Marrs, F. Thomson, P. Cowley, A. Christopoulos, R. G. Pertwee and R. A. Ross, Mol. Pharmacol., 2005, 68, 1484–1495 CrossRef CAS PubMed.
- D. A. Jackson and S. A. Mabury, Environ. Toxicol. Chem., 2009, 28, 1866 CrossRef CAS PubMed.
- B. Hoelke, S. Gieringer, M. Arlt and C. Saal, Anal. Chem., 2009, 81, 3165–3172 CrossRef CAS PubMed.
- M. M. C. van D. Lee, M. Bras, C. J. van Koppen and G. J. R. Zaman, J. Biomol. Screening, 2008, 13, 986–998 CrossRef PubMed.
- B. D. Kangas, M. S. Delatte, V. K. Vemuri, G. A. Thakur, S. P. Nikas, K. V. Subramanian, V. G. Shukla, A. Makriyannis and J. Bergman, J. Pharmacol. Exp. Ther., 2013, 344, 561–567 CrossRef CAS PubMed.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- Organisation for Economic Co-operation and Development, Test No. 117: Partition Coefficient (n-octanol/water), HPLC Method, OECD Publishing, Paris, 2004 Search PubMed.
- X. Fei and Q. Zheng, J. Liq. Chromatogr. Relat. Technol., 2005, 28, 939–945 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra01212g |
| This journal is © The Royal Society of Chemistry 2014 |