
Synthesis, characterization, and solution structure of all-conjugated polyelectrolyte diblock copoly(3-hexylthiophene)s
Abstract
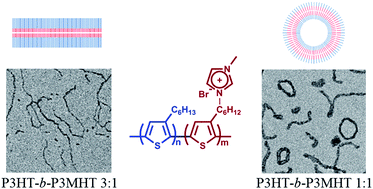
We report on a new class of all-conjugated polyelectrolyte diblock copoly(3-hexylthiophene)s, poly(3-hexylthiophene)-b-poly[3-[6-(N-methylimidazolium)hexyl]thiophene] (P3HT-b-P3MHT). The polyelectrolyte diblock copolymers were prepared by a catalyst-transfer Kumada polycondensation followed by quaternization of the bromohexyl side groups of the poly[3-(6-bromohexyl)thiophene] block with N-methylimidazole. The obtained diblock copolymers have well-defined block ratios, narrow polydispersity indices, and high regioregularity. Their characterization as well as the thermal, crystalline, and optical properties, and self-assembly behavior have been investigated in detail. Transmission electron microscopy (TEM) provided evidence for solvent-induced self-assembly. A series of morphologies including short or long nanowires and nanorings could be obtained depending on the selectivity of solvents toward different blocks and the block ratios.
 Please wait while we load your content...
Please wait while we load your content...

