
Short and efficient synthesis of a daunosamine donor from l-fucal†
Abstract
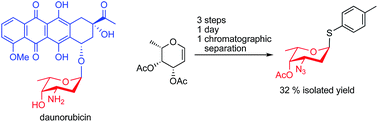
Anthracyclines, e.g. daunorubicin, doxorubicin, and idarubicin, consist of a tetracycline moiety linked via a glycosidic bond to a sugar residue, usually the aminosugar daunosamine. The anthracyclines are efficient chemotherapeutic agents against cancer, but their use is limited due to cardiotoxicity and induction of multidrug resistance. In the search for new anthracycline analogs, a daunosamine donor that can be used to glycosylate suitable aglycons is of utmost importance. Here, we present a short and efficient synthesis of the versatile donor p-tolyl 4-O-acetyl-3-azido-2,3,6-trideoxy-1-thio-α-L-lyxo-hexopyranoside in 3 steps from commercially available L-fucal with an overall yield of 32%. The same procedure can be used to synthesize the donor p-tolyl 4-O-acetyl-3-azido-2,3,6-trideoxy-1-thio-α-L-arabino-hexopyranoside in 28% overall yield from L-rhamnal, for the synthesis of epirubicin analogs.
 Please wait while we load your content...
Please wait while we load your content...

