
Simultaneous determination of a broad range of cardiovascular drugs in plasma with a simple and efficient extraction/clean up procedure and chromatography-mass spectrometry analysis
Abstract
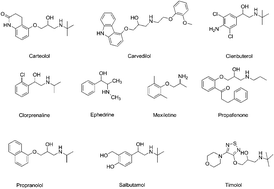
A simple, easy to use and efficient method was described for simultaneous determination of ten cardiovascular drugs with a broad range of physicochemical properties in rat plasma via online solid phase extraction (online SPE) and high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). Following a simple centrifugation step, a 10 μL aliquot of the plasma sample was injected directly onto the HPLC system. The LiChrospher® RP-18 ADS (25 mm × 4 mm, 25 μm, Merck) cartridge was washed with 10 mM ammonium acetate buffer (pH 9.5) for 1 min, after which time the analytes were removed by back-flushing directly onto the analytical column (Acclaim 120 C18 column, 150 mm × 4.6 mm, 5 μm) with gradient elution using acetonitrile–10 mM ammonium acetate buffer (pH 3.5) as mobile phase. The flow rate through both columns was 1 mL min−1, and the analytes were quantified using a triple-quadrupole tandem mass spectrometer in multiple-reaction monitoring mode. Linear calibration curves were obtained over the range of 0.2–100 ng mL−1, and the method limits ranged from 0.2 ng mL−1 to 1 ng mL−1 which is sensitive enough for clinical drug monitoring. The intra- and inter-day precisions were in the range of 0.20–2.32%, and the accuracies were between 93.33% and 114.60%. Excellent recoveries from plasma were achieved with a range from 83.52% to 107.38%. The procedure was easier to execute and required less sample handling than methods previously described in the literature. This easy to use and high-throughput method with direct injection of plasma samples for the analysis of multiple cardiovascular drugs may provide a practical solution for tailoring drug dosage in a rational manner to rapidly achieve optimal efficacy and safety of medication.
 Please wait while we load your content...
Please wait while we load your content...

