
Role of graphite precursor and sodium nitrate in graphite oxide synthesis†
Abstract
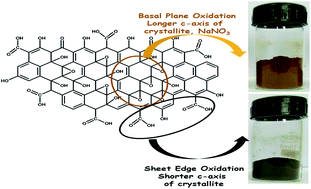
Graphite oxides were synthesized following the Hummer's method using three different graphite precursors. The role of the graphite precursor and sodium nitrate during graphite oxide synthesis has been investigated. Different analytical techniques, namely powder X-ray diffraction, thermal gravimetric analysis, infrared spectroscopy, and ultraviolet-visible absorption spectroscopy have been implemented to characterize the synthesized graphite oxides. The study revealed that a longer c-axis (axis perpendicular to the carbon layer) in the graphite crystallite favored basal plane oxidations over sheet edge oxidations on the graphitic sheets during graphite oxide synthesis. These basal plane oxidations caused a strain in the graphitic sheets. Due to this strain, graphitic layers were cracked along the a-axis (axis in the carbon layer planes) and also layers were peeled off along the c-axis. Hence, crystallite sizes of the synthesized graphite oxides were significantly reduced compared to their graphite precursors. Furthermore, basal plane oxidations and consequently reduction in the crystallite sizes were enhanced by the addition of sodium nitrate during the synthesis.
 Please wait while we load your content...
Please wait while we load your content...

