Mechanism of N-heterocyclic carbene-catalyzed chemical fixation of CO2 with aziridines: a theoretical study†
Weiyi Li*a,
Dongfeng Huangb and
Yajing Lva
aResearch Center for Advanced Computation, School of Physics and Chemistry, Xihua University, Chengdu, Sichuan 610039, P. R. China. E-mail: weiyili@mail.xhu.edu.cn; Fax: +86-028-87648043; Tel: +86-028-87648043
bCollege of Chemistry and Chemical Engineering, Anyang Normal University, Anyang, Henan 455000, P. R. China
First published on 2nd April 2014
Abstract
The reaction mechanism of cycloaddition of CO2 with N-benzylaziridine catalyzed by N-Heterocyclic Carbenes (NHCs) has been investigated using density functional theory (DFT) at the M06-2X (IEFPCM, 2-propanol)/6-311++G(d,p)//M06-2X/6-31G(d,p) level. The calculations reveal that the reaction prefers to proceed through a three step mechanism mediated by free NHC rather than being catalyzed by the NHC–CO2 adduct. Free NHC plays a role as the catalyst precursor to promote the initial ring-opening of the aziridine with the incorporation of CO2 through SN2 anti nucleophilic attack, leading to the formation of the carboxylate intermediate. Then, the generated carboxylate as an active intermediate can easily react with the excess of N-benzylaziridine and CO2. Finally, the intramolecular nucleophilic addition allows the release of the cycloaddition product with the recovery of the active intermediate. Compared with background reaction, the higher nucleophilicity of free NHC as well as the stabilization from the t-Bu group on the nitrogen atom of the imidazolium ring help to lower the energy barrier of the ring-opening step, which accelerates the formation of the active intermediate and suppresses the generation of by-product oligomer. In addition, the calculations predict that the NHCs bearing the additional ring fusion beside the C–C and C–N bonds of the imidazolium ring might be more powerful catalysts for chemical fixation of CO2 with aziridines, owing to the enhancement of the nucleophilicity of the NHCs and the reactivity of the carboxylate intermediate.
1. Introduction
As we all know, carbon dioxide (CO2) is one of the major greenhouse gases in the atmosphere, which leads to global warming. On the other hand, CO2 is also one of the most easily available renewable carbon resources, which has the advantages of being nontoxic, abundant, inexpensive and non-flammable.1 Therefore, the utilization of CO2 as C1 building block in synthesis chemistry has becomes a significant and challenging research subject that could contribute not only to the mitigation of the concentration of CO2 in atmosphere, but also towards the economic and environmental friendly synthesis of value-added products, such as oxygen-containing compounds,2 nitrogen-containing compounds,3 C–C unsaturated hydrocarbons,4 and so on.However, due to the thermodynamic stability and kinetic inertness of CO2, highly reactive species, like small-membered epoxides and aziridines, are usually employed as the reagents in CO2 chemical fixation. In recent years, much effort has been directed to catalytic incorporation of CO2 into epoxides with the formation of the useful cyclic carbonate products.5 Aziridines, as the analogues of epoxides, are also the high active candidate that can react with CO2 (Scheme 1). The resulting oxazolidones are very important 5-membered heterocyclic compounds, which exhibits wide applications as chiral auxiliaries and synthetic intermediates in several asymmetric transformations.6 Hence, the capture of CO2 with aziridines to afford oxazolidones has been widely studied, and numerous homogenous and heterogeneous catalytic systems have successfully been developed for this cycloaddition reaction.
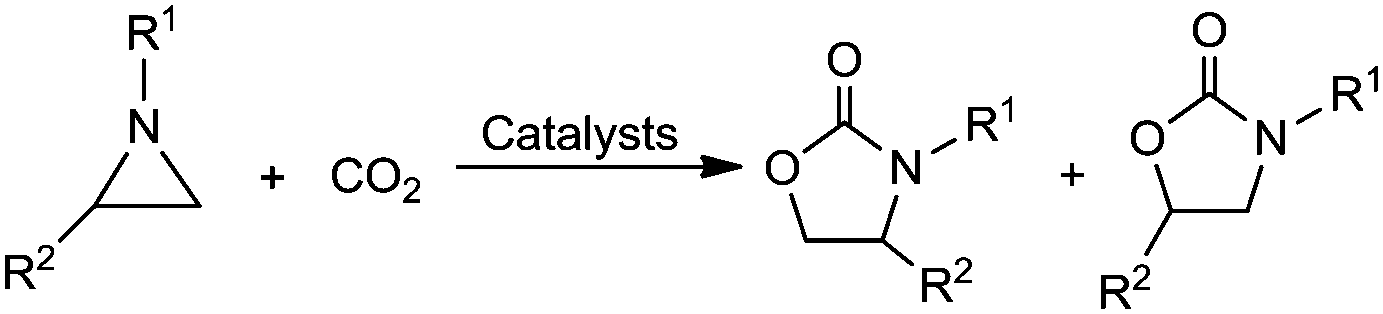 | ||
| Scheme 1 Catalytic chemical fixation of CO2 with aziridines to oxazolidones. | ||
In early research, dual-component system, viz., I2/scCO2,7a phenol/DMAP,7b salenCr(III)/DMAP,7c alkali metal halide (LiI, LiBr etc.),7d,e tetra-alkylammonium halide,7f organic bases (DBU and DBN),7g and n-Bu2SnO7h have been explored for the chemical fixation CO2 with aziridines. In most of the these catalytic cases, toxic organic solvents or additives, high pressure of CO2 or supercritical CO2 system, high catalyst loading or low reusability of catalysts, long reaction time, or a combination of these features are generally required to achieve oxazolidones with high yields. Although some significant advantages and green catalytic systems, such as ZrOCl2·8H2O,8a 1,4-diazabicyclo[2.2.2]octane (DABCO)-based Lewis basic ionic liquids,8b naturally occurring α-amino,8c polyethylene glycol functionalized phosphonium salt (Br−Ph3+PPEG600P+Ph3Br−),8d protic onium salts,8e and even under catalyst-free and solvent-free conditions,8f have been made for this reaction, however, the reaction scopes are mostly limited to the substrates in which the 2-positon carbon atom and nitrogen atom are substituted with the aromatic groups. In addition, oligomeric and polymeric as by-products were inevitably generated in many reaction systems.
N-Heterocyclic carbenes (NHCs), with their lone pair of carbene electrons, have received considerable attentions as nucleophiles, ligands for transition metals, building blocks in heterocyclic construction and organocatalysts in a number of synthetic transformations.9 Especially, it is well known that NHCs show good affinity to CO2.10 The resulting imidazolium carboxylates (NHC–CO2 adducts) are extensively established as a good and convenient CO2 carrier. In NHC–CO2 adducts, it is commonly accepted that CO2 moiety is activated by the lone pair of carbene electrons, which serves as oxidant and nucleophile in subsequent transformations. However, in most of the reactions it is difficult to identify the catalytically active species is free NHC or NHC–CO2 adduct. The catalytic role of free NHC or NHC–CO2 is also in controversial.11 For instance, Zhang and co-workers11a reported NHC-catalyzed the reduction of CO2 to methanol with silanes as the hydrogen source, and proposed that NHC was the catalytically active species, which enhanced the reactivity of CO2 is via the formation of NHC–CO2 adduct. On the contrary, Wang and co-workers11b investigated the mechanism of such reaction by using DFT method. Their calculation shows that the catalytic role of NHC is in the activation of Si–H bonds of silanes rather than the activation for CO2. The oxidation reaction of aromatic aldehydes to aromatic carboxylic acids with CO2 as oxidation over NHC catalyst was studied by Zhang11c and Nair11d group, respectively. Zhang et al. suggested that the initial activation of CO2 by free NHC is followed by the nucleophilic attack of NHC–CO2 adduct to the aldehyde. However, Nair and co-workers supposed that the first step of the reaction is the addition of NHC to the aldehyde, which subsequently reacted with CO2. Ren et al.11e performed a theoretical study on the mechanism of this catalytic reaction at B3LYP/6-31G(d,p) level. Two different activation modes were calculated and compared, which indicated that the activation of the aldehydes by the free NHC is slightly energy-favorable. In addition, the catalytic transformation of CO2 with epoxides has been realized by NHCs, and the reaction mechanism was investigated by experimental and theoretical methods. Lu and co-workers10c studied thermal stability of NHC–CO2 adducts by means of in situ FTIR method and analyzed the N-substituent effect on the electron density over the imidazolium ring. Ajitha and Suresh12 explored the reaction mechanism at MPWB1K/6-311++G(3df,2p) level of theory. The calculations revealed that the catalytically active species was free NHC rather than NHC–CO2 adduct.
Very recently, Ikariya and co-workers13 reported that the recyclable imidazolium 2-carboxylates derived from NHCs and CO2 was efficient to promote cycloaddition of CO2 with tertiary aziridines bearing various substituents on the nitrogen atom, which gave the target product oxazolidones with 92% yield (Scheme 2). However, the reaction was carried out under a harsh experimental condition (5.0 MPa, 363 K), and a small amount of the undesired oligomeric by-products was concomitantly obtained. Although two possible reaction mechanisms, corresponding to NHC–CO2 adduct and aziridines–CO2 zwitterionic mediated catalytic cycles were proposed, the actual catalytic component as well as the precise mechanism remains yet to be uncertain. These findings motivated us to perform a comprehensive mechanistic study on such reaction by means of DFT calculations, aiming to identify the catalytically active species and design more efficient NHC catalysts so that the chemical fixation of CO2 with aziridines can be taken under mild condition with high yield and selectivity to the desired oxazolidones.
 | ||
| Scheme 2 Cycloaddition of N-benzylaziridine (1a) with CO2 to 3-benzyl-2-oxazolidone catalyzed by NHC–CO2 adducts. | ||
2. Computational details
The hybrid meta exchange-correlation function M06-2X, developed by Zhao and Truhlar,14 was demonstrated to outperform the normal function (e.g. B3LYP) in handling main group thermochemistry, kinetics, and noncovalent interactions. Accordingly, the geometries of the reactants, products, intermediates (IMs), and transition states (TSs) in the present system were fully optimized by M06-2X method with 6-31G(d,p) basis set.15 In order to assess the sensibility of the results to basis sets, the geometries of the key IMs and TSs were re-optimized at M06-2X/6-311++G(d,p) level. This benchmark indicates that the geometrics and relative energies of the species calculated at the two levels are quite close to each other (see in ESI†). The vibrational frequency were calculated on the same level to characterize the nature of the stationary points as true minima (with no imaginary frequency) or transition states (with unique imaginary frequency), and obtain zero-point vibrational energy (ZPE) and thermal corrections. Intrinsic reaction coordinates (IRC)16 were also used to confirm the transitions states correctly connect the corresponding minima.To consider the solvent effect, single-point energies of all the species in 2-propanol (experimentally used) were calculated at M06-2X/6-311++G(d,p) level by employing IEFPCM17 solvent model. The free energies in the solvent (Gsol) were obtained by the combination of these single-point energies with Gibbs free energy corrections in gas phase. However, because such thermal corrections are based on the ideal gas-phase model, entropy contributions to free energies for reactions in solvent medium are inevitably overestimated.18 In particular for reactions involving component changes, the suppressing effect of the solvent on the rotational and transitional freedoms of the substrates is usually ignored. Martin et al.19 have proposed to correct the overestimation of entropic contribution by artificially elevating the reaction pressure from 1 atm to 1354 atm. This protocol was applied by Wang and co-workers in the theoretical study on the mechanism of CO2 reduction reaction.20 According to their approach, a correction of 4.3 kcal mol−1 applies per component change for a reaction at 298 K and 1 atm [i.e., a reaction from m to n components has an additional correction of (n − m) × 4.3 kcal mol−1]. The free energies corrected by Martin et al.'s approach were used in the following discussion.
Furthermore, Natural Bond Orbital (NBO)21 analysis was performed on the optimized-structures to obtain a further insight into the electronic and chemical bond properties of the system. The nucleophilicity index N22,23 of the reactants, intermediates and catalytic active species was also performed by computing the HOMO and LUMO energies at the ground-state of the molecules involved. All theoretical calculations were carried out with Gaussian 09 programs.24 The computed structures were drawn using CYLVIEW program.25
3. Results and discussion
3.1 Reaction of CO2 and N-benzylaziridine without NHC–CO2 adduct
In order to explore the catalytic role of NHC–CO2 adduct, the reaction mechanism of cycloaddition CO2 with N-benzylaziridine in the absence of the NHC–CO2 adduct was first investigated in the present work. Three possible pathways, presented in Scheme S1 (in ESI†), are discussed to show the efforts to discover the minimum energy reaction pathway (MERP), which is extracted from Scheme S1,† and redrawn in Scheme 3. The potential energy profile is shown in Fig. 1, together with the optimized structures of the transition states involved. | ||
| Scheme 3 The energy-favorable reaction pathway for cycloaddition of 1a and CO2 without NHC–CO2 adduct. | ||
 | ||
| Fig. 1 Potential energy profile for cycloaddition of 1a and CO2 without NHC–CO2 adduct along the MERP. The bond distances of the optimized structures are given in Å. | ||
Along the MERP, the reaction begins from the formation of ternary complex 6, in which CO2 is weakly interacted with the nitrogen atom of 1a. The formation of this complex is exothermic by 2.6 kcal mol−1 in enthalpy but unfavorable by 11.0 kcal mol−1 (after free energy correction) in free energy because of entropy penalty. From complex 6, the SN2 type aziridine ring-opening can take place through transition state TS6-7, leading to the generation of zwitterionic intermediate 7. At transition state TS6-7, the nitrogen atom of 1a as a nucleophile attacks the carbon atom of 1a from the backside of the leaving nitrogen atom, while CO2 as a Lewis acid is inserted to the leaving nitrogen atom with the construction of C–N bond. With respect to the separated reactants (1a + CO2), a high energy barrier of 36.0 kcal mol−1 is required for the ring-opening step. From the zwitterionic intermediate 7, the intramolecular nucleophilic attack of the oxygen atom of carboxylate moiety on the carbon atom happens via transition state TS7-2 with the free energy barrier of 19.3 kcal mol−1 relative to 7. Downhill from transition state TS7-2, the target product 3-benzyl-2-oxazolidone 2 can be yielded with the regeneration of 1a simultaneously. Hence, the substrate 1a can be regarded as the catalyst for this cycloaddition process, which plays the roles as both nucleophile for ring opening and Lewis base for the incorporation of CO2 fixation. On the other hand, it can be found that the positive aziridine or negative carboxylate moiety of intermediate 7 might subsequently react with an external 1a or the couple of 1a and CO2, affording intermediate 8 or 9. Transition state TS7-8 with free energy barrier of 26.4 kcal mol−1 is 6.0 kcal mol−1 preferred than transition state TS7-9, and comparable with transition state TS7-2 in free energy, suggesting the generation of intermediate 8 is probable in kinetics. Once intermediate 8 is formed, it may continue to react with 1a and CO2, resulting in oligomeric by-products. The calculation accords well with the experimental result that the low yield of cycloaddition product was obtained, accompanied with undesired oligomeric by-products in the absence of NHC–CO2 adduct.13
3.2 Reaction of CO2 and N-benzylaziridine catalyzed by NHC–CO2 or free NHC
Next, the reaction mechanism in the presence of NHC–CO2 adduct was studied. As mentioned in the literatures,10 NHC–CO2 adduct is not very thermodynamically stable. Under high temperature and the existence of epoxides, the decomposition of NHC–CO2 adduct can be occurred with the release of the free NHC and CO2, especially for NHC–CO2 adduct bearing large steric substituents on the nitrogen atom of the imidazolium ring. Both NHC–CO2 adduct and the released free NHC might be the catalytic active component for the chemical fixation of CO2. To study the reaction mechanisms of cycloaddition of CO2 and 1a catalyzed by NHC–CO2 adduct 10 and free NHC 11, the structure and activity of these two catalytic species was initially compared.As shown in Fig. 2, in the zwitterionic NHC–CO2 adduct 10, the molecular electrostatic potential (MESP) analysis26 shows a strong charge separation in this complex, the imidazolium ring is positively charged while the two terminal oxygen atoms of carboxylate moiety is negatively charged. NBO analysis indicates that the carbene lone pair is donated to the carbon atom of CO2, leading to the activation of CO2 molecule, as evidenced by a decrease in the Wiberg bond index of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from 1.896 in free CO2 to 1.515. Compared with free CO2, the global nucleophilicity index (N) of 10 is increased from −1.5 eV to 2.8 eV (Table 1). On the other hand, in free NHC 11, the electron-rich region is predominately focused on the carbene center. The global nucleophilicity index of 11 is calculated to be 3.6 eV, indicating that the nucleophilicity of 11 might be stronger. Since both 10 and 11 are competent as the nucleophile to react with the electrophile 1a and CO2, two different catalytic reaction pathways were calculated to identify the actual active species. The detailed free energy profiles are provided in Fig. 3.
O from 1.896 in free CO2 to 1.515. Compared with free CO2, the global nucleophilicity index (N) of 10 is increased from −1.5 eV to 2.8 eV (Table 1). On the other hand, in free NHC 11, the electron-rich region is predominately focused on the carbene center. The global nucleophilicity index of 11 is calculated to be 3.6 eV, indicating that the nucleophilicity of 11 might be stronger. Since both 10 and 11 are competent as the nucleophile to react with the electrophile 1a and CO2, two different catalytic reaction pathways were calculated to identify the actual active species. The detailed free energy profiles are provided in Fig. 3.
 | ||
| Fig. 2 The visualization of the MESP mapped on to the van der Waals surface, HOMO orbital for NHC–CO2 adduct and free NHC. | ||
| Species | μ [a.u.] | η [a.u.] | ω [eV] | N [eV] |
|---|---|---|---|---|
| CO2 | −0.46 | 0.45 | 6.3 | −1.5 |
| 1a | −0.30 | 0.29 | 4.1 | 2.8 |
| 10 | −0.32 | 0.28 | 4.8 | 2.8 |
| 11 | −0.27 | 0.26 | 3.8 | 3.6 |
| 17 | −0.32 | 0.29 | 4.9 | 2.5 |
| 22 | −0.28 | 0.21 | 4.9 | 4.2 |
 | ||
| Fig. 3 Potential energy profile for cycloaddition of 1a and CO2 catalyzed by NHC–CO2 adduct and free NHC, respectively. | ||
For NHC–CO2 adduct 10 catalyzed reaction, three possible reaction pathways were located (see Scheme S2 in ESI†). For a concise expression, the energy-preferable catalytic cycle is illustrated in Scheme 4. Stating from 10, the initial formation of ternary complex 16, is followed by a concerted aziridine ring-opening and CO2 insertion. In complex 16, the external CO2 is strongly interacted with the nitrogen atom of 1a, which polarizes and weakens the C–N bond of 1a, as an evidence of a decreased Wiberg bond index (from 0.965 to 0.884). Similar to complex 6, the binding energy is preferable by 2.0 kcal mol−1 in enthalpy but unfavorable in free energy owing to the entropy loss. Then, one oxygen atom of carboxylate moiety attacks the carbon atom of 1a through SN2 anti nucleophilic addition. Transition state TS16-17 relates to the simultaneous breaking of the C–N bond of 1a and the bonding of CO2 molecule with the formation of a new C–N bond, which requires an energy barrier of 34.0 kcal mol−1 relative to the separated reactants and 10. During the ring-opening of 1a with the incorporation of CO2, the evolution of the electronic population along the reaction path is analyzed. Fig. 5a displays the evolution of charge on the NHC–CO2 moiety, nitrogen atom of 1a and the external CO2 as the reaction proceeds along the IRC. From complex 16 to intermediate 17, the positive charge on the NHC–CO2 moiety increases gradually, while the negative charge is accumulated on the external CO2 moiety. The change of the charge on the nitrogen atom is smooth, indicating the nitrogen atom serves as the bridge for the charge transfer from the NHC–CO2 moiety to the carboxylate moiety. Compared with 1a itself promoted ring-opening process, although the global nucleophilicity index of 10 is not significantly increased (N = 2.8 eV), however, the relative Gibbs free energy of TS16-17 is 2.0 kcal mol−1 lowered than that of TS6-7. This might be attributed to the extra stabilizing interactions between the oxygen atom of the carboxylate moiety and the two hydrogen atoms of t-Bu groups on the imidazolium ring (Fig. 4). This weak interaction helps the delocalization the accumulated negative charge the carboxylate moiety and favor the aziridine ring-opening. In the following step, intermediate 17 can be converted to the isomer 18 via TS17-18, a conformational change transition state for the C–N bond rotation (ΔG≠ = 7.0 kcal mol−1). Owing to the breakage of the weak interactions, this process is endothermic by 0.9 kcal mol−1 in free energy. Finally, intermediate 18 undergoes an intermolecular nucleophilic attack of the oxygen atom of the carboxylate moiety on the carbon atom of the methylene via five-membered-ring transition state TS18-10, allowing the formation of cycloaddition product 2 and the regeneration of the catalyst 10.
 | ||
| Scheme 4 The most energy-favourable reaction pathways for cycloaddition of 1a and CO2 catalyzed by NHC–CO2 adduct. | ||
 | ||
| Fig. 4 The optimized structures (bond distance in Å) of the key transition states in cycloaddition of 1a and CO2 catalyzed by NHC–CO2 adduct. | ||
Furthermore, as the reaction was carried out under excess CO2 with high pressure (5.0 MPa),13 the possibility of another termolecular reaction pathway is considered wherein intermediate 17 subsequently reacts with one more couple of 1a and CO2. However, the calculation shows that the nucleophilic attack from the terminal oxygen atom of the carboxylate moiety to the carbon atom of 1a via transition states TS19-20 should be kinetically not allowed, as the relative free energy of TS19-20 is as high as 44.7 kcal mol−1 with respect to the zero point. The reason might be the intermediate 17 is thermodynamically unstable and bears the weaker nucleophilicity (N = 2.5 eV). Overall, for NHC–CO2 adduct 10 catalyzed cycloaddition of 1a with CO2, the ring-opening transition state TS16-17 with the highest energy requirement (HER) of 34.0 kcal mol−1 should be turnover frequency determining transition state (TDTS)27 on the MERP. Relative to 1a-promoted ring-opening, the catalytic effect of 10 is 2.0 kcal mol−1 in free energy.
Subsequently, reaction mechanism catalyzed by free NHC was investigated (Scheme 5). In this case, free NHC 11 is initially formed via TS10-11, a transition state for the decarboxyaltion of 10. The free energy barrier of this process is 15.5 kcal mol−1, indicating that the formation of free NHC 11 is available in kinetics. The calculation is in agreement with the experimental results reported by Louie and co-workers that NHC–CO2 adduct 10 loses CO2 and decomposes at lower temperature (344 K).10b In the presence of 11, the ring-opening of 1a takes places from ternary complex 21 through transition state TS21-22 to give the zwitterionic intermediate 22. At TS21-22, the carbene center of 11 approaches the carbon atom of 1a from the backside of the leaving nitrogen atom to promote the cleavage the C–N bond of 1a, while CO2 is bonded to the nitrogen center with the construction of a new C–N bond. During this process, there is also a charge transfer from the free NHC to the carboxylate moiety (Fig. 5b). The magnitude of charge variation for 11-mediated ring-opening is sharper than that of 10-promoted one, meaning that the charge transfer is more facile. Similarly, with the aid of the stabilizing interactions from the hydrogen atoms of t-Bu group substituted on the nitrogen atom of the imidazolium ring (Fig. 6), the increased negative charge on the carboxylate moiety can be effectively delocalized. Relative to the separated reactants (10 + 1a + CO2), the overall barrier of 11-mediated ring-opening step is 32.6 kcal mol−1, which is lower than the ones in 1a and 10-promoted reaction pathways. The computed result is compatible with the fact that the nucleophilicity of free NHC 11 is stronger than those of 1a and NHC–CO2 adduct. Downhill from TS21-22, the negative charge accumulated on the carboxylate moiety in intermediate 22 (−0.659e) is smaller than that in intermediate 17 (−0.694e). As a result, intermediate 22 is thermodynamically more stable than 17. From intermediate 22, the intermolecular nucleophilic attack from the oxygen atom in the carboxylate moiety to the carbon atom of the methylene can be occurred via the ring-closing transition state TS22-11, which leads to the production of 2 and the recovery of 11. The calculation predicts the Gibbs free energy barrier of this step is 37.3 kcal mol−1, and the completion of the catalytic cycle is exothermic by 12.8 kcal mol−1.
 | ||
| Scheme 5 The catalytic cycles for cycloaddition of 1a and CO2 catalyzed by free NHC. | ||
 | ||
| Fig. 5 Evolution of the charge populations in ring-opening of 1a with the insertion of CO2 catalyzed by NHC–CO2 adduct (a) or free NHC (b). | ||
 | ||
| Fig. 6 The optimized structures (bond distance in Å) of the key transition states in cycloaddition of 1a and CO2 catalyzed by free NHC. | ||
Alternatively, the stable intermediate 22, with the negative charge accumulated on the carboxylate moiety, has the stronger nucleophilicity (N = 4.2 eV) and is capable to react with the excess CO2 and 1a. From ternary complex 23, the ring-opening of 1a via transition state TS23-24 permits the yield of another stable intermediate 24. NBO analysis shows an analogue charge transfer from the carboxylate moiety to the external CO2 molecule. The negative charge on the carboxylate moiety decreases from −0.681e to −0.280e, while the negative charge on the external CO2 increases from −0.385e to −0.687e. Relative to intermediate 22, the free energy barrier of this ring-opening step is 27.2 kcal mol−1, which is relatively lower than the corresponding ones in the other pathways. Finally, the production of 2 and the regeneration of active intermediate 22 can be achieved through the analogue intermolecular ring-closing transition state TS24-22 by overcoming low free energy barrier of 16.5 kcal mol−1.
On the basis of the above computed results, it can be found that the ring-opening of 1a with the accompanying insertion of an external CO2 to the nitrogen atom of 1a is rate-determining for 1a, NHC–CO2 adduct and free NHC-mediated cycloaddition reaction. Among these three catalytic processes, free NHC-promoted ring-opening of 1a via transition state TS21-22 with the lowest energy barrier of 32.7 kcal mol−1 is energetically more favored. Free NHC plays the role as the catalytic precursor to trigger the formation of active intermediate 22. Once active intermediate 22 is generated, the subsequent ring-opening of 1a with the fixation of CO2 catalyzed by 22 is more facile (ΔG≠ = 27.2 kcal mol−1). Intermediate 22 and transition state TS23-24 serves as the turnover frequency intermediate (TDI) and turnover frequency transition state (TDTS), respectively, controlling the turnover frequency (TOF)27 of the catalytic cycle. The calculation is similar to the theoretical result of NHC-catalyzed CO2 fixation with epoxide reported by Suresh and co-worker,12 and well accounts for the experimental observations that the NHC catalyst can suppress the formation of undesired oligomeric.13
3.3 Catalytic effect of other NHCs
The understanding of the reaction mechanism motivated us to inspect the catalytic activity of more free NHCs, as the experiment was carried out under a relatively harsh condition (363 K 5.0 MPa).13 The previous theoretical study on the electronic and steric properties of the various free NHCs suggested that the electron-donating groups (–NMe2, –OMe) substituted on the C4 and C5 of the imidazolium ring can enhance the electro-rich character of the carbene lone pair.28 The ring fusion at the C–C and C–N bonds of the imidazolium ring may influence the electronic effect on the carbene center. To compare the catalytic effect between 11 and other NHCs, more free NHCs were selected to catalyze the cycloaddition reaction of 1a with CO2 in the present theoretical simulation (Scheme 6). The calculated nucleophilicity indexes of the free NHCs as well as the corresponding activation free energies in the two ring-opening steps are summarized in Table 2. | ||
| Scheme 6 The selected free NHCs in the cycloaddition of 1a and CO2. | ||
| System | N | ΔG≠1 | ΔG≠2 |
|---|---|---|---|
| 11 | 3.6 | 32.6 | 27.2 |
| NMe-11 | 4.1 | 32.0 | 29.6 |
| OMe-11 | 3.9 | 32.9 | 28.5 |
| Triazol-5-ylidene | 3.2 | 34.3 | 28.1 |
| Thiazol-2-ylidene | 3.2 | 32.2 | 27.3 |
| ImBicar | 3.7 | 28.9 | 26.9 |
| ImCylm | 3.9 | 26.6 | 29.2 |
| ImDpylm | 4.7 | 29.3 | 26.9 |
| ImPhen | 3.6 | 28.8 | 24.2 |
The calculations show that when electron-donating (–NMe2, –OMe) are introduced at the C4 and C5 atoms of NHC ring, the nucleophilicity of free NHC NMe-11 and OMe-11 is stronger than free NHC 11. The result is in line with the theoretical investigation that the electron-donating group substituted at the C-positions of NHC is effective for making an electron-rich carbene center.28 However, when these two free NHCs are used as the nucleophiles to promote the ring-opening of 1a with the insertion of CO2, the Gibbs activation free energies (ΔG≠1) are not decreased. The reason might due to the large repulsion between the substituents at C-positions (–NMe2 and –OMe groups) and N-positions (t-Bu group) of NHC ring, leading the structures of the transition states unstable. Hence, free NHCs NMe-11 and OMe-11 might not perform better catalytic effect than 11 in the chemical fixation of CO2 with aziridines. Additionally, triazol-5-ylidene and thiazol-2-ylidene, as the commonly used NHC organocatalysts, the catalytic effect of them were also evaluated. The calculations predict that the nucleophilicity of these two free NHCs are inferior to free NHC 11, and thereby the energy barriers in the two ring-opening steps are slightly higher. These two kinds of free NHCs seem to be less effective for cycloaddition of CO2 with aziridines as well. The satisfactory result appears when the saturated/unsaturated ring fusion was introduced at the C–C and C–N bonds of the imidazolium ring. In the cases of these four NHCs, the free energy barriers in the first ring-opening step significantly fall with the increased global nucleophilicity, suggesting that these four free NHCs might be in favor of accelerating the rate of the formation active intermediate. The saturated fused-ring NHCs ImCylm performs best catalytic effect (ΔG≠1 = 26.6 kcal mol−1). However, for the second ring-opening step, the free energy barrier in the catalytic system of ImCylm are 2.0 kcal mol−1 higher than that in free NHC 11, suggesting that TOF of the catalytic cycle might be decreased in this catalytic system. When bipyridine-derived NHC ImPhen is employed as the catalyst precursor, it not only exhibits the comparable catalytic efficiency with free NHC ImBicar and ImDpylm in the first ring-opening step, but also a better the catalytic performance in the second ring-opening step (ΔG≠2 = 24.2 kcal mol−1). This might be due to the higher activity of the resulting carboxylate intermediate. As a result, this free NHC is predicted to be the more effect catalysts for the present reaction system. The introduction of the additional ring fusion beside the C–C and C–N bonds of the imidazolium ring is either in the advantage of enhancing the electron-rich character of the carbene center or the reactivity of the active intermediate, which might be helpful for the chemical fixation of CO2 with aziridines.
4. Conclusions
The mechanism of the chemical fixation of CO2 with N-benzylaziridine catalyzed by NHC has been theoretically investigated using DFT method at M06-2X (IEFPCM, 2-propanol)/6-311++G(d,p)//M06-2X/6-31G(d,p) level. The major conclusions are listed as follows:The calculations confirm that the catalytic active species is free NHC rather than NHC–CO2 adduct. Free NHC plays the role as the catalyst precursor to trigger the ring-opening of the aziridine with the insertion of CO2, leading to the formation of the active intermediate. Compared with the reaction catalyzed by the substrate N-benzylaziridine, the energy barrier of the ring-opening step mediated by free NHC is decreased from 36.0 to 32.6 kcal mol−1, owing to the higher nucleophilicity of free NHC as well as the stabilization from the t-Bu group substituted on the nitrogen atom of the imidazolium ring. Once the active intermediate is generated, it can easily react with the excess of N-benzylaziridine and CO2 by overcoming the lower free energy barrier of 27.2 kcal mol−1, which selectively gives the desired cycloaddition product and suppresses the generation of the by-product oligome.
Furthermore, the catalytic effect of more free NHCs is theoretically evaluated. The calculations predict that the introduction of ring fusion at the C–C and C–N bonds of the imidazolium ring may either enhance the electron-rich character of the carbene center or the reactivity of the active intermediate. The fused-ring NHCs might be the more powerful catalysts for the chemical fixation of CO2 with aziridines.
Acknowledgements
The authors are grateful for the financial support from the Key Scientific Research Found of Xihua University (no. Z1313319) and the Scientific Research Fund of Education Department of Sichuan Province (no. 14ZB0131).References
- For reviews see:
(a) T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS
; (b) S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347 RSC
-
(a) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC
- Z. Yang, L. He, J. Gao, A. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602 CAS
-
(a) I. Omae, Catal. Today, 2006, 115, 33 CrossRef CAS
-
(a) X. Lu, W. Ren and G. Wu, Acc. Chem. Res., 2012, 45, 1721 CrossRef CAS
-
(a) G. Zappia, E. Gacs-Baitz, G. D. Monache, D. Misiti, L. Nevola and B. Botta, Curr. Org. Synth., 2007, 4, 238 CrossRef CAS
-
(a) H. Kawanami and Y. Ikushima, Tetrahedron Lett., 2002, 43, 3841 CrossRef CAS
-
(a) Y. Wu, L. He, Y. Du, J. Wang, C. Miao and W. Li, Tetrahedron, 2009, 65, 6204 CrossRef CAS
-
(a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed
-
(a) Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., Int. Ed., 2009, 48, 4194 CrossRef CAS PubMed
-
(a) S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322 CrossRef CAS PubMed
- M. J. Ajitha and C. H. Suresh, Tetrahedron Lett., 2011, 52, 5403 CrossRef CAS
- A. Ueno, Y. Kayaki and T. Ikariya, Green Chem., 2013, 15, 425 RSC
-
(a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS
-
(a) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS
-
(a) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS
-
(a) W. Sang-Aroon and V. Ruangpornvisuti, Int. J. Quantum Chem., 2008, 108, 1181 CrossRef CAS
-
(a) C. Zhang, R. Zhang, Z. Wang, Z. Zhou, S. Zhang and Z. Chen, Chem.–Eur. J., 2009, 15, 5910 CrossRef CAS PubMed
- R. L. Martin, P. J. Hay and L. R. Pratt, J. Phys. Chem. A, 1998, 102, 3565 CrossRef CAS
-
(a) F. Huang, C. Zhang, J. Jiang, Z. Wang and H. Guan, Inorg. Chem., 2011, 50, 3816 CrossRef CAS PubMed
-
(a) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS
-
(a) L. R. Domingo and J. A. Sáez, Org. Biomol. Chem., 2009, 7, 3576 RSC
- The global electrophilicity index ω,22c,d which measures the stabilization energy when the system acquires an additional electronic charge ΔN from the environment, is given in terms of the electronic chemical potential μ and chemical hardness η by the following simple expression:22e ω[eV] = (μ2/2η). Both quantities can be calculated in terms of the HOMO and LUMO electron energies, εH and εL, as μ ≈ (εH + εL)/2 and η ≈ (εH − εL), respectively. The nucleophilicity index N,22b based on the HOMO energies obtained within the Kohn–Sham scheme, is defined as N = EHOMO(Nu) − EHOMO(TCE).22b The nucleophilicity is taken relative to tetracyanoethylene (TCE) as a reference, because it has the lowest HOMO energy in a large series of molecules already investigated in the context of polar cycloadditions.
- M. J. Frisch, et al., Gaussian 09, Reversion A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, Sherbrooke, Québec, Canada, 2009, http://www.cylview.org Search PubMed
-
(a) P. Politzer and D. G. Truhlar, Chemical applications of atomic and molecular electrostatic potentials: reactivity, structure, scattering, and energetics of organic, inorganic, and biological systems, Plenum Press, New York, 1981 Search PubMed
-
(a) S. Kozuch and S. Shaik, J. Am. Chem. Soc., 2006, 128, 3355 CrossRef CAS PubMed
- M. J. Ajitha and C. H. Suresh, J. Org. Chem., 2012, 77, 1087 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Computational methods, energies, optimized geometries and the full citation of Gaussian 09 program. See DOI: 10.1039/c4ra01018c |
| This journal is © The Royal Society of Chemistry 2014 |