
Catalyst-free synthesis of cycloalkenyl phosphonates†
Abstract
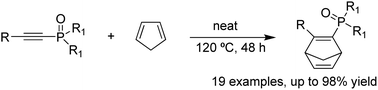
The reactions described provide a facile and efficient access to cycloalkenyl phosphonates with good to excellent yields via Diels–Alder cycloadditions between alkynyl phosphonates and 1,3-dienes under catalyst-free conditions.
 Please wait while we load your content...
Please wait while we load your content...

