The molecular dynamics of different relaxation modes in asymmetric chlorinated butyl rubber/petroleum resin blends
Fengshun Zhang,
Guansong He,
Kangming Xu,
Hong Wu* and
Shaoyun Guo*
The State Key Laboratory of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Chengdu 610065, China. E-mail: wh@scu.edu.cn; nic7702@scu.edu.cn; Fax: +86-28-85466077; Fax: +86-28-85405135
First published on 24th April 2014
Abstract
The relaxation behavior and different modes of molecular motion in the miscible blends of asymmetric chlorinated butyl rubber (CIIR) and petroleum resin (PR) were investigated by dynamic mechanical spectroscopy (DMS) and dielectric spectroscopy (DS). The different modes of CIIR molecular motion, attributed to local segmental motion, sub-Rouse modes and Rouse mode relaxation, have been detected from both of the DMS and DS results, and the relaxation times of local segmental motion and Rouse modes could be fitted by the Vogel–Fulcher–Tammann equation. Due to the increased activation energy with increasing PR content, local segmental motion of CIIR is slightly confined, however, Rouse modes of CIIR, which contain more backbone bonds and need a larger free volume than the local segmental motion, are greatly confined. As a result, the relaxation temperature of local segmental motion moves slightly to high temperature but that of Rouse modes moves to high temperature more significantly.
1. Introduction
In the past decades, the glass transition temperature (Tg), which is the most significant and important transition in amorphous polymers from the glass to the rubber state, has attracted much interest in condensed matter physics.1–5 The corresponding studies on polymers will benefit from understanding the mechanisms of various relaxation processes, which are important in both basic research and technological applications of this important class of materials.6 However, the dynamic behavior (the glass transition) of polymer is very complex and includes different processes extending over a huge time/frequency range and also covering different length scales. The studies7–10 have shown that different relaxation processes, local segmental motion (or glass transition), sub-Rouse modes and Rouse modes relaxation processes, are responsible for the glass transition region (or the softening dispersion), which is important to study the nature of molecular mobility in polymers near the glass transition region. The local segmental motion is the correlated motion of local segments which determines the glass transition (Tg), while the sub-Rouse modes and the Rouse modes should be attributed to the motion of longer chain segments by Ngai, Plazek and co-workers.11–13 The local segmental motion, which contains 2 or 3 backbone bonds, needs small free volume, while sub-Rouse modes and the Rouse modes, which contain in the order of 50 or more backbone bonds, need larger free volume than the local segmental motion.Although the different modes of molecular motion mentioned in the above are present in the softening dispersion region of all amorphous polymers, it is difficult to resolve the contribution of each mode from one another due to overlap, and not all of them are necessarily observed together in an experiment using one technique.14,15 Only in the case of individual polymers, like polyisobutylene (PIB)16 and its derivatives:17 butyl rubber (a copolymer of isobutylene and isoprene, IIR) and chlorinated butyl rubber (a chlorinated copolymer of isobutylene and isoprene, CIIR), which have very broad softening dispersion, has the sub-Rouse modes been resolved from the Rouse modes by dynamic mechanical measurements,8 dielectric spectroscopy9 and dynamic light scattering.12 As reported in the literatures, these processes have very different temperature and frequency dependences.
It has also attracted the high interest how the dynamics of PIB and its derivatives are influenced by changing the local conditions of molecular chains. In the PIB/polystyrene (PS) blends, Wu and coworkers18 have chosen PS with higher glass transition temperature to form space confinement on the PIB phase. The temperature positions of different relaxation modes are reduced with decreasing particle size of the PIB phase, and the depression amplitude of Rouse modes is larger than that of local segmental motion.19 However, the size of the confined space, which is about several micrometers in diameter due to the poor miscibility between PS and PIB, is much larger than that of the average molecular chain.20 Furthermore, they have also studied the miscible PIB blends with a plasticizer and find that the addition of the plasticizer does not reduce the intermolecular coupling but the effective chain packing is disrupted. As a result, the Rouse modes come closer to the sub-Rouse modes and local segmental relaxation, the softening dispersion becomes narrower, and eventually the shoulder disappears. As known, the miscible polymer blends, whose components have very different Tg, can be considered as dynamic asymmetric systems.21 Researches indicate that the dynamics of one component in the dynamic asymmetric blends can be different from that of its neat polymer. Moreover, the observed changes can serve as critical tests of theories and models of polymer dynamics and viscoelasticity constructed primarily for homopolymers.22,23 Nevertheless, the PS can not confine the relaxation at molecular length scales, and the plasticizer promotes rather than confines the molecular motion of PIB. This inspires us to study how the different modes of polymer are confined by another miscible polymer with higher relaxation temperature.
It also has been reported that the CIIR is widely applied and researched because of high vulcanization speed and well dielectrically active compared with PIB.17 In addition, the CIIR is miscible with petroleum resin (PR) and form a single-phase amorphous blend, the relaxation process of CIIR/PR blends shifts to higher temperature with increasing PR content.24,25 Therefore, the CIIR/PR blend, whose components have different Tg, can be investigated as dynamic asymmetric systems.22,23 Nevertheless, little information has been reported about the relaxation mechanism of this asymmetric blend. In the present work, the aim of incorporating slow component (PR) into CIIR is to make confinement effect on the motion of fast component (CIIR) molecular chains, and dynamic mechanical spectroscopy (DMS) is used alongside dielectric spectroscopy (DS) to provide a plausible interpretation of the different relaxation dynamics in the blends.
2. Experimental
2.1. Materials
The chlorinated butyl rubber (CIIR, product code: CBK 139, Tg = −63.5 °C) with a chlorination concentration of 1.2% was from Nizhnekamsk (Russia). The petroleum resins (PR, product code: A2100, Tg = 51.4 °C) synthesized from aliphatic C5 petroleum feedstreams was from Puyang Ruisen Petroleum Resins Factory, China. The stearic acid was purchased from Kelong Co. (Chengdu, China). All these materials were used as purchased.The number average molecular weight of CIIR is 6.955 × 105 g mol−1 with a polydispersity of 1.74. The number average molecular weight of PR is 2.237 × 104 g mol−1 with a polydispersity of 4.1. The information mentioned above regarding the molecular weight and its polydispersity were determined by gel permeation chromatography (GPC, AGILENT-1100) in tetrahydrofuran at 25 °C. The Tg mentioned above was tested by differential scanning calorimetry.
2.2. Preparation of samples
A laboratory-scale mixer equipped with a pair of roller blades was used for blending. Blends of CIIR with PR and stearic acid (HSt) content 98.5/0/1.5, 88.5/10/1.5, 78.5/20/1.5, 68.5/30/1.5 (CIIR/PR/HSt, in wt%) were prepared via mechanical mixing at room temperature for 8 min, then the blends were compression molded to form sheets with 2 mm in thickness under a pressure of 10 MPa for 10 min at 130 °C. The polar small molecular HSt was used as the dielectric probes to improve the dielectric response character of weak polarity CIIR, which was beneficial to monitor local segmental motion, sub-Rouse modes and Rouse modes in the dielectric spectra.17,262.3. Characterization
Dynamic mechanical spectroscopy (DMS) was measured by TA Q800 (TA company, USA) analyzer under a heating rate of 3 °C min−1 within a temperature range of −80 °C to 100 °C and a frequency of 10 Hz for temperature scanning. The samples were trimmed to dimensions of 15 mm in length, 10 mm in width and 2 mm in thickness. The tan delta (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) is defined by the ratio loss (E′′) and storage modulus (E′). The E′′ represents the viscous component of the modulus and includes all the energy dissipation processes during dynamic strain, and the E′ represents the stiffness component of the modulus. The tanδ, which can be used to measure the vibration energy dissipation, is also known as loss factor.24,25
δ) is defined by the ratio loss (E′′) and storage modulus (E′). The E′′ represents the viscous component of the modulus and includes all the energy dissipation processes during dynamic strain, and the E′ represents the stiffness component of the modulus. The tanδ, which can be used to measure the vibration energy dissipation, is also known as loss factor.24,25
Dielectric spectroscopy (DS) measurement was performed on a Novocontrol GmbH Concept 80 dielectric spectrometer in the frequency range from 10−1 to 107 Hz. During the measurement the sample was contained between two parallel plates (diameter 23 mm, gap 2 mm). Temperature was controlled using a nitrogen-gas cryostat with temperature stability better than 0.1 °C.
Differential scanning calorimetry (DSC) test was conducted on a Q20 calorimeter (TA Instruments). The sample was cooled to −80 °C from room temperature at a cooling rate of 10 °C min−1 and stabilized for 5 min, and then heated at a heating rate of 10 °C min−1 to 80 °C.
3. Results and discussion
3.1. Dynamic mechanical spectroscopy (DMS)
DMS has been extensively used to detect the change of molecular dynamics of polymers.11 The tanδ curves of CIIR and CIIR/PR blends at 10 Hz are presented in the Fig. 1. Generally, The Tg values of CIIR can be extracted from the peak temperatures in the tanδ curves. However, it can be observed that the tanδ curve of CIIR displays an asymmetrical peak with a shoulder at about −33 °C and a maximum at −13 °C. On the basis of literatures,6,8,9,11,12 the tanδ peak can be resolved into three peaks corresponding to local segmental, sub-Rouse modes and Rouse modes peaks respectively. The local segmental, which is a cooperative motion of neighboring chains each involve several repeat units, is enthalpic and determines the glass transition temperature. Meanwhile, the Rouse modes, which is based on the motions of Gaussian submolecules formed by sufficient number of repeat units in each chain, are entropic in nature.27 From Fig. 1, it can be observed that both of the shoulder and the maximum of the tanδ peak of CIIR move to the higher temperature due to the incorporation of PR, indicating that the slow component (PR) with high glass transition at 51 °C, forms space confinement on the fast component (CIIR) molecular chains. It is interesting that the space confinement affects the local segmental motion and Rouse modes relaxation processes to different extents. Due to the presence of PR molecules, local segmental motion of CIIR is slightly confined, however, the Rouse modes of CIIR is greatly confined (as show in Fig. 2a). As a result, the shape of the tanδ peak is significantly altered with increasing PR content in the blends, moreover, the distance of the maximum and the shoulder becomes wider and wider. It is known that the temperature of the relaxation peak partly reflects energy barriers in the rotation and reptation of molecular chains, and the high temperature indicates the high energy barrier. In this case, the Rouse modes of confined CIIR require more energy than that of unconfined one, therefore, the maximum of the loss peak move towards high temperature and more mechanical vibration energy is converted to heat energy. As a result, from Fig. 2b, it can be noticed that the loss peak height of local segmental motion is dramatically suppressed with decreasing PR content in the blends, but that of Rouse modes is little changed. Thus, PR plays a role like anti-plasticizer in determining different transition temperatures and increasing the energy barrier to confine the mobility of CIIR chains.
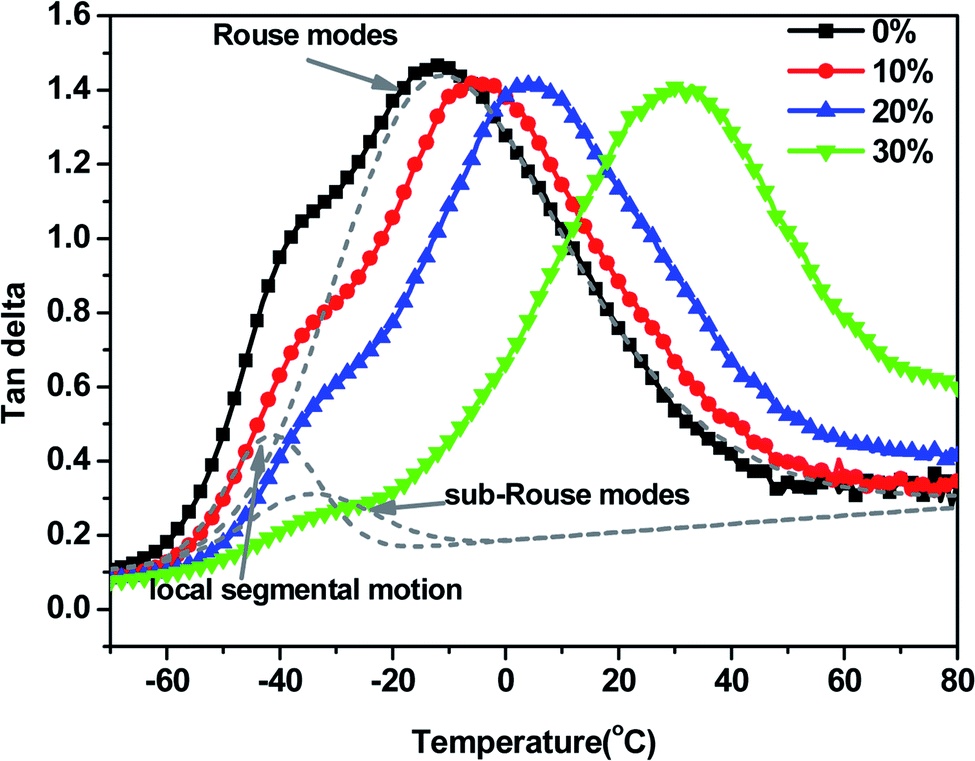 | ||
| Fig. 1 tanδ versus temperature curves of blends with different PR content. The dashed gray lines are the fitting of local segmental motion, sub-Rouse modes and Rouse modes peaks of neat CIIR, respectively. | ||
 | ||
| Fig. 2 Effect of PR content on the peak height (a) and peak position (b) of CIIR. | ||
3.2. Dielectric spectroscopy (DS)
Broadband dielectric relaxation spectroscopy is used to interrogate the dynamics of the softening dispersion in CIIR over a broad frequency and temperature range.8,15,16 The study of polymers as a function of frequency and temperature can be used to elucidate the effects of intermolecular co-operative motions and hindered dielectric rotations.15,28 Generally, the dielectric loss spectra correspond to the Havriliak–Negami (HN) susceptibility function for most polymers.29
 | (1) |
Typical frequency and temperature dependence of DS spectra of CIIR are shown in Fig. 3a, and two dielectric loss peaks are clearly observed in all dielectric spectra. The relaxation time (τ) is defined as (2πfmax)−1, where fmax refers to the frequency corresponding to the maximum of the dielectric loss peak. The larger peak at high frequency with short relaxation time is attributed to local segmental motion, and the small peak at low frequency with long relaxation time is attributed to the Rouse modes. In the middle-frequency range, sub-Rouse modes main contribute to the stretched region on the low frequency side of the local segmental motion peaks. It should be noted that the precise location of sub-Rouse peak is difficult to pinpoint, but the experimental data significantly deviate from the HN equation due to sub-Rouse relaxation. As shown in the Fig. 3b, each curve of dielectric loss vs. frequency displays an asymmetric double-peak structure with a shoulder on the low-frequency side and a maximum on the high-frequency side, attributing to the relaxation of local segmental motion, sub-Rouse modes and Rouse modes of CIIR, respectively. No evidence for the loss peak of PR can be detected due to its very weak dielectric activity, while the decrease in the relaxation strength is associated with the incorporation of PR, which has weak dipoles and low dielectric loss.
 | ||
| Fig. 3 Representative dielectric spectra of as a function of the CIIR (a) and CIIR/PR blend with 30% PR content (b) frequency at different temperatures. | ||
In order to understand more precisely the molecular interactions between CIIR and PR, especially the effect of PR on the different modes of molecular motion of CIIR, the relaxation times of local segmental motion and Rouse modes are plotted in Fig. 4. It can be observed that both local segmental motion and Rouse modes shift to higher temperature. Moreover, local segmental motion and Rouse modes have different temperature and frequency dependences.2,26
 | ||
| Fig. 4 Temperature dependence of τLSM and τR for CIIR and CIIR/PR blend with 30% PR content. | ||
These relaxation processes are identified as segmental thermally activated processes in origin, generally, the peak shifts to lower frequency as temperature decreases with a relaxation time following an Arrhenius type equation:
|
τ = τ0exp(H/kT)
| (2) |
The temperature dependence of relaxation time for local segmental motion and Rouse modes seems to follow better the Vogel–Fulcher–Tammann (VFT) equation:17
|
τ = τ0exp(B/(T − T0))
| (3) |
| log(τ0) | B | T0 (K) | Coefficient of determination | TDMS (K) | TDSC (K) | TDS (K) | |
|---|---|---|---|---|---|---|---|
| Local segmental motion of CIIR | −12.56 | 1077.8 | 129.7 | 0.998 | 239.9 | 209.5 | 203.73 |
| Rouse modes of CIIR | −6.40 | 475.4 | 188.5 | 0.998 | 262.3 | — | 245.15 |
| Local segmental motion of CIIR/PR blend | −12.36 | 961.4 | 145.9 | 0.999 | 244.2 | 217.7 | 212.88 |
| Rouse modes of CIIR/PR blend | −0.11 | 3.7 | 276.3 | 0.997 | 293.2 | — | 278.03 |
 | ||
| Fig. 5 Temperature dependence of heat flow curves of CIIR and CIIR/PR blend with 30% PR content. | ||
4. Conclusions
In this work, the motion of the fast component in the softening dispersion is strongly confined by the slow component. As the two relaxation processes need different free volume, the local segmental motion and Rouse modes have different responses to the space limited from both DMS and DS data. The PR plays a role like anti-plasticizer in decreasing the free volume fraction of the blend. The presence of PR molecules slightly confines the molecular mobility of the local segmental motion of CIIR, but greatly confines the molecular mobility of Rouse modes of CIIR.Acknowledgements
Financial supports of the National Natural Science Foundation of China (51273132, 51227802 and 51121001) and Program for New Century Excellent Talents in University (NCET-13-0392) are gratefully acknowledged.References
- J. E. Pye and C. B. Roth, Phys. Rev. Lett., 2011, 107, 2357011–2357015 CrossRef.
- M. Song, X. Y. Zhao, Y. Li, S. K. Hu, L. Q. Zhang and S. Z. Wu, RSC Adv., 2014, 4, 6719–6729 RSC.
- S. Arrese-Igor, A. Alegría, A. J. Moreno and J. Colmenero, Macromolecules, 2011, 44, 3611–3621 CrossRef CAS.
- J. Colmenero and A. Arbe, Soft Matter, 2007, 3, 1474–1485 RSC.
- D. J. Plazek and K. L. Ngai, Physical Properties of Polymers Handbook, ed. E. M. James, Springer, New York, 2007, vol. 1, pp. 187–215 Search PubMed.
- D. J. Plazek, I. C. Chay, K. L. Ngai and C. M. Roland, Macromolecules, 1995, 28, 6432–6436 CrossRef CAS.
- X. B. Wu and Z. G. Zhu, J. Phys. Chem. B, 2009, 113, 11147–11152 CrossRef CAS PubMed.
- X. A. Wang, G. S. Huang, J. R. Wu, Y. J. Nie and X. J. He, J. Phys. Chem. B, 2011, 115, 1775–1779 CrossRef CAS PubMed.
- M. Paluch, S. Pawlus, A. P. Sokolov and K. L. Ngai, Macromolecules, 2010, 43, 3103–3106 CrossRef CAS.
- X. A. Wang, G. S. Huang, J. R. Wu, Y. J. Nie, X. J. He and K. W. Xiang, Appl. Phys. Lett., 2011, 99, 1219021–1219023 Search PubMed.
- D. J. Plazek, X. D. Zheng and K. L. Ngai, Macromolecules, 1992, 25, 4920–4924 CrossRef CAS.
- A. K. Rizos, K. L. Ngai and D. J. Plazek, Polymer, 1997, 38, 6103–6107 CrossRef CAS.
- P. G. Santangelo, K. L. Ngai and C. M. Roland, Macromolecules, 1993, 26, 2682–2687 CrossRef CAS.
- K. L. Ngai, Eur. Phys. J. E: Soft Matter Biol. Phys., 2002, 8, 225–235 CrossRef CAS PubMed.
- S. H. Zhang, X. Jin, P. C. Painter and J. Runt, Macromolecules, 2002, 35, 3636–3646 CrossRef CAS.
- K. Kunal, M. Paluch, C. M. Roland, J. E. Puskas, Y. Chen and A. P. Sokolov, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1390–1399 CrossRef CAS.
- J. R. Wu, G. S. Huang, X. A. Wang, X. J. He and J. Zheng, Soft Matter, 2011, 7, 9224–9230 RSC.
- J. R. Wu, G. S. Huang, X. A. Wang, X. J. He and B. Xu, Macromolecules, 2012, 45, 8051–8057 CrossRef CAS.
- J. R. Wu, G. S. Huang, X. A. Wang, X. J. He and H. X. Lei, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 2165–2172 CrossRef CAS.
- B. Imre, K. Renne and B. Pukánszky, eXPRESS Polym. Lett., 2014, 8, 2–14 CrossRef CAS.
- K. L. Ngai and S. Capaccioli, J. Chem. Phys., 2013, 138, 0549031–05490319 Search PubMed.
- A. J. Moreno and J. Colmenero, Phys. Rev. Lett., 2008, 100, 1260011–1260014 Search PubMed.
- V. A. Harmandaris, K. Kremer and G. Floudas, Phys. Rev. Lett., 2013, 110, 1657011–1657015 CrossRef.
- C. Li, G. Z. Wu, F. Y. Xiao and C. F. Wu, J. Appl. Polym. Sci., 2007, 106, 2472–2478 CrossRef CAS.
- C. Li, S. A. Xu, F. Y. Xiao and C. F. Wu, Eur. Polym. J., 2006, 42, 2507–2514 CrossRef CAS PubMed.
- K. Kessairi, S. Napolitano, S. Capaccioli, P. Rolla and M. Wübbenhorst, Macromolecules, 2007, 40, 1786–1788 CrossRef CAS.
- D. J. Plazek, C. A. Bero, S. Neumeister, G. Floudas, G. Fytas and K. L. Ngai, Colloid Polym. Sci., 1994, 272, 1430–1438 Search PubMed.
- C. V. Chanmal and J. P. Jog, eXPRESS Polym. Lett., 2008, 2, 294–301 CrossRef CAS.
- J. S. Havriliak and S. J. Havriliak, Polymer, 1996, 37, 4107–4110 CrossRef.
- X. B. Wu, S. Y. Shang, Q. L. Xu, C. S. Liu, Z. G. Zhu and G. Z. Zhang, Appl. Phys. Lett., 2008, 93, 0119101–0119103 Search PubMed.
- J. Swenson, G. A. Schwartz, R. Bergman, W. S. Howells and J. Swenson, Eur. Phys. J. E, 2003, 12, 179–183 CrossRef CAS PubMed.
- D. Fragiadakis, R. Casalini, R. B. Bogoslovov, C. G. Robertson and C. M. Roland, Macromolecules, 2011, 44, 1149–1155 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |