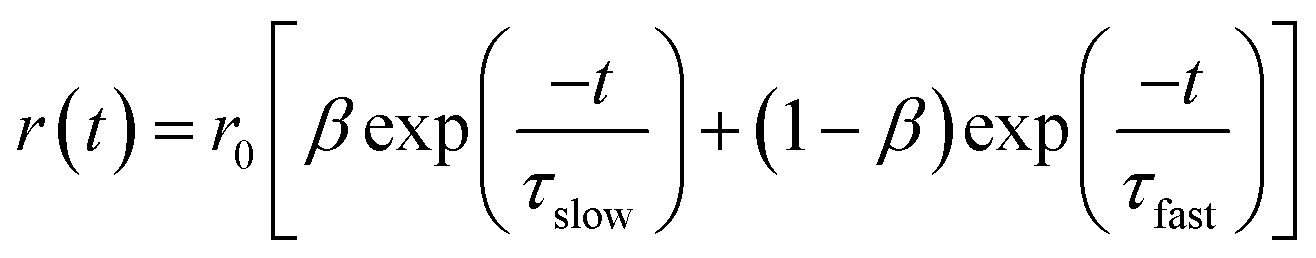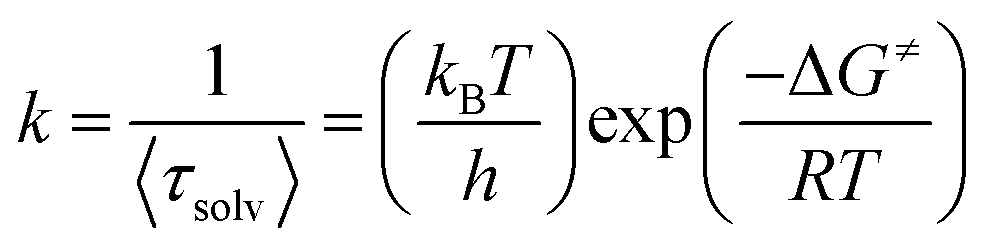DOI:
10.1039/C4RA00965G
(Paper)
RSC Adv., 2014,
4, 13989-14000
Influence of double confinement on photophysics of 7-(diethylamino)coumarin-3-carboxylic acid in water/AOT/isooctane reverse micelles†
Received
3rd February 2014
, Accepted 4th March 2014
First published on 4th March 2014
Abstract
The effect of double confinement on the photophysics of 7-(diethylamino)coumarin-3-carboxylic acid (7-DCCA) inside the water pool of water/AOT/isooctane reverse micelles has been reported in this study. At first a supramolecular host–guest complex was formed in water between 2-hydroxypropyl-γ-cyclodextrin (HP-γ-CD) and 7-DCCA. Then the aqueous solution of this complex was used to form reverse micelles at any particular w0 value (w0 = [water]/[surfactant]). We have used sodium dioctyl sulfosuccinate (AOT) as surfactant and isooctane as non-polar solvent to prepare reverse micelles. A comparative study between double confinement system and the single confinement system, where the 7-DCCA molecule was incorporated inside the core of the water/AOT/isooctane reverse micelles, was done. We have used the steady state absorption and fluorescence emission techniques to highlight the significant shift of the spectral behaviour of the 7-DCCA due to the double encapsulation of the dye in the nanopool of the reverse micelles. More affirmation has been achieved by the use of time resolved fluorescence emission spectroscopy. The study of solvation dynamics and rotational relaxation dynamics was used as tools to investigate the effect of double encapsulation on the excited state dynamics of the probe molecule. These excited state dynamics clearly show that even at the highest w0 value studied here, the excited state dynamics of the doubly confined dye are significantly different from those of the single confined dye in the reverse micelle. The higher values of fluorescence emission decay time, rotational relaxation and solvent relaxation times in the doubly confined system compared to those of the single confined system at different w0 values proved the existence of the supramolecular host–guest complex inside the core of the water/AOT/isooctane reverse micelle.
1. Introduction
Coumarin has been a very popular class of compound; due to its excellent optical properties it has been used in several photophysical studies.1–3 Substituted amino coumarins have been used as popular fluorescence probe molecules in several studies due to their sensitivity to the environment.4–7 The photophysical properties of 7-aminocoumarins are dependent on the polarity and viscosity of the media and on the specific solute solvent interaction with the media. These substituted amino coumarin dyes form an intramolecular charge transfer (ICT) state when excited in a polar medium. After the formation of ICT state, the di-substituted amino group undergoes twisting to produce highly polar twisted intramolecular charge transfer (TICT) state.8–11 This TICT state formation is essentially a nonradiative process. These TICT states may be emissive or non emissive in nature. These coumarin dyes have been popularly used to study solvation dynamics in neat solvents.12–14 The solvation dynamics in different confined medium like micelles, reverse micelles etc. are much slower than that in neat solvent, due to the restriction imposed on the solvent molecules in these medium. Here in this study we have used reverse micelle as the confined medium. Reverse micelles are the aggregates of surfactant molecules in nonpolar solvents, where the polar or ionic head groups of the surfactants point towards the polar solvent pool and the long nonpolar hydrocarbon chain pointing towards the nonpolar solvents.15,16 Aqueous reverse micelles facilitate the solubilisation of sufficient amount of water in the core.17,18 Water/AOT/isooctane reverse micelle is a ternary system composed of water, Aerosol OT (AOT) surfactant and isooctane (oil). The water molecules trapped inside the core of reverse micelle are excellent model of biological water.19–23 The water molecules present in the water pool of the reverse micelles are different in behaviour compared to bulk water. Several techniques have been employed for studying the structural features of AOT reverse micelles.24–31 Gorski and Ostanevich using the small-angle neutron scattering reported several important features of AOT reverse micelles.32,33 All the water molecules in the core, except six water molecules per AOT molecule freeze at −40 °C.27 Jain et al.34 by FT-IR studies showed that three different kind of water molecules are present inside the core of reverse micelle and these are trapped water, bound water and free water. They described that the trapped water molecules are that water molecules which reside at the interface between the polar head groups of the surfactants and do not bind to any other groups and molecules. They demonstrated that the trapped water molecules are matrix isolated and monomeric in nature. The bound water molecules are generally the water molecules, which are strongly hydrogen bonded to the polar head groups of the AOT surfactants through sodium counter ions and are held tightly. This layer is somewhat immobilised layer. The last category of water molecules inside the pool comprises of free water molecules, these are strongly hydrogen bonded. They represented that with the gradual addition of water to the reverse micelle core the number of bound water molecules per AOT surfactant molecule increased regularly upto w0 ≈ 18, beyond this it remained constant. The number of free water molecule increases gradually upto w0 = 10 and this increase continues to be constant in between w0 = 10 and 18. Beyond w0 ≈ 18, further addition of water to the pool causes the increase in the number of free water molecules. Extensive studies have been made by several groups on coumarin molecules in reverse micelles.35–40 The solvent relaxation of water in reverse micelle systems are slower than the bulk water.
In this study we have showed the formation of double confinement of a molecule in the reverse micelles and effect of double confinement on the photophysics of 7-(diethylamino)coumarin-3-carboxylic acid (7-DCCA). Double confinement means initially dye-2-hydroxypropyl-γ-cyclodextrin (HP-γ-CD) complex in water was formed, and then this host–guest complex in water was injected in the water/AOT/isooctane reverse micelles. The water solution of host–guest complex will be in the core of the reverse micelles due to very low solubility of dye and HP-γ-CD in isooctane. As a result of the double confinement of the dye, restrictedness on the dye is increased and this leads to the improvement of the fluorescence emission properties. We have used the dye 7-DCCA as being hydrophilic, it has great propensity to get dissolved to a significant extent in water, unlike other amino coumarins, which have very low solubility in water. So when trapping inside the core of reverses micelle, 7-DCCA and 7-DCCA-HP-γ-CD complex is expected to reside entirely inside the water pool of the reverse micelle. Previously, Jane et al. have studied the double confinement of Coumarin 522 inside the host–guest supramolecular complex with β-cyclodextrin in a water/AOT/n-heptane reverse micelle.41 Coumarin 522 was more hydrophobic than 7-DCCA. 7-DCCA is highly hydrophilic due to the presence of hydrophilic acid group and at the same time is a TICT forming dye. We have used 2-hydroxypropyl-γ-cyclodextrin (HP-γ-CD) because of its high solubility in water42 compared to normal γ-CD to ensure complete solubilisation of the supramolecular host guest complex inside the water pool of water/AOT/isooctane reverse micelles. HP-γ-CD shows greater affinity for complex formation than γ-CD.42 In this study we are showing the double confinement of a molecule in the reverse micelles and its effect on the emission properties of the molecule. This study will help us to understand the modification and improvement of photophysical properties of a hydrophilic and TICT forming dye (like 7-DCCA) upon a double confinement in the reverse micelles. For this purpose we have used the fluorescence emission properties of 7-DCCA as tool. The change in emission quantum yields, solvent relaxation, rotational relaxation of 7-DCCA upon double confinement was used as experimental evidences. Previously very few studies are reported regarding the photophysical properties of this 7-DCCA dye.43–46 We have studied the effect of single confinement on the photophysical properties of 7-DCCA in the nano-pool of water/AOT/isooctane reverse micelles of different sizes and reported the excitation wavelength dependent photophysics. We had also demonstrated the red edge excitation shift in the core of reverse micelle.47 In this study we have shown the double confinement of a hydrophilic molecule 7-DCCA in the core of the reverse micelles and its effect on the fluorescence emission properties. We have compared our result with our previous study,47 where we had demonstrated the effect of single confinement on the photophysics of 7-DCCA in the reverse micelles.
2. Materials and methods
7-DCCA was purchased from Sigma-Aldrich and used as received. Sodium dioctylsulfosuccinate (AOT) was purchased from Sigma-Aldrich. AOT was dried under vacuum and used as previously described in literature. For all the experiments the concentration of AOT has been maintained at 0.1 (M). Isooctane was purchased from Spectrochem, India. The required amount of water has been added to the system to form the reverse micelles at different w0 values, where w0 is represented as [water]/[AOT]. 2-Hydroxypropyl-γ-cyclodextrin (HP-γ-CD) was purchased from Sigma-Aldrich and used as received. For the preparation of aqueous solution of 7-DCCA and reverse micelles Millipore water was used. For the double confinement of the molecule, we have used the procedure described by Jane et al.41 For the host guest complexation in neat water, the concentration of 7-DCCA and HP-γ-CD was maintained at 5.5 × 10−4 (M) and 6 × 10−2 (M). During the double confinement studies the dye concentration is maintained at 3 × 10−6 (M). The final concentration of HP-γ-CD during this double confinement studies is maintained at 3.2 × 10−4 (M). UV-Vis absorption studies were carried out using UV-Vis spectrophotometer (model: UV-2550, Shimadzu). The steady state fluorescence emission measurements were executed using a Fluoromax-4P spectrofluorometer (Horiba Jobin Yvon). In the case of steady state absorption and fluorescence measurement the temperature was kept constant at 298 K by using a Jeiotech refrigerated bath circulator (model: RW0525G). The fluorescence quantum yields of 7-DCCA + HP-γ-CD complex in different reverse micellar media were measured using the fluorescence quantum yield of Coumarin 480 in water solution (ϕf = 0.66) as reference,48 by using the following equation:| | 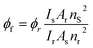 | (1) |
where s and r stand for the sample and reference respectively. Here I stands for the integrated area under the fluorescence curve, A stands for the absorbance of the sample at excitation wavelength and n stands for the refractive index of the medium.
The fluorescence time resolved decays were measured by using a picosecond time-correlated single-photon counting (TCSPC) technique. We have used a time-resolved fluorescence spectrophotometer from Edinburgh Instruments (model: LifeSpec-II, U.K.) for lifetime measurement. We have used picosecond diode lasers with excitation wavelength at 405 nm. The full width at half maximum (FWHM) of our system is ∼90 ps. The fluorescence transients were detected at magic angle (54.7°) polarization using Hamamatsu MCP PMT (3809U) as a detector. The decays were analysed by using F-900 decay analysis software. The fluorescence anisotropy decay (r(t)) was measured by using the same instrument. The following equation was used to obtain r(t).
| | 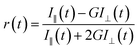 | (2) |
where the emission intensities at parallel (
I‖) and perpendicular (
I⊥) polarizations were collected alternatively by fixing the time for both the decays. We had used motorised polarizers to collect the parallel and perpendicular decays.
G is the correction factor for the detector sensitivity to the polarization direction of the emission. Similar method was used to measure the
G factor. F-900 software was used to analyze the anisotropy decay.
The overall anisotropy decay is represented by the following equation which is the sum of two exponential equations:49
| | 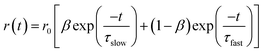 | (3) |
where
r0 is the limiting anisotropy,
β represents the relative contribution of the slow component and (1 −
β) represents the relative contribution of the fast component. The slow and the fast components are denoted by
τslow and
τfast. The average rotational relaxation time is represented by the following equation:
49| | | 〈τrot〉; = βτslow + (1 − β)τfast | (4) |
For the time resolved measurements the temperature was kept constant at 298 K by using Peltier-controlled cuvette holders from Quantum Northwest (model: TLC-50).
In order to study the solvation dynamics we have constructed time resolved emission spectra (TRES) according to the procedure described in the literature.50,51 The spectrum at any time (t) is represented as S(λ; t) and is describe as follows:
| | 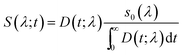 | (5) |
where
D(
t;
λ) is representative of fitted decays. Thus
S(
λ;
t) can be easily obtained from the fitted decays
D(
t;
λ) by the relative normalisation of the steady state emission spectrum. All TRES are fitted by using the log-normal function:
| | 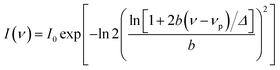 | (6) |
where
νp,
I0,
Δ, and
b stand for peak frequency, peak height, width parameter and asymmetric parameter, respectively. To get the solvent relaxation time we have deduced the solvent response function [
C(
t)]:
| | 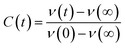 | (7) |
ν(
t),
ν(0),
ν(∞) are the peak frequencies at the time
t, at the time
t = 0 and at
t = ∞. Now, the decay of
C(
t) with time can be fitted by the exponential function:
| | 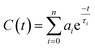 | (8) |
where
ai stands for the amplitude and τ
i stands for the solvent relaxation time constant. The average solvation time can be expressed using the following equation:
| | 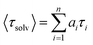 | (9) |
3. Result and discussion
3.1 Steady state absorption and emission spectral studies
7-DCCA is an acidic dye remaining in the water solution as a deprotonated species.45 It shows a prominent and single absorption peak at 409 nm in water. We have studied the steady state absorption spectral feature of double confined 7-DCCA in the CD/water/AOT/isooctane reverse micelles and compared this result with the single confined system. We had reported earlier the spectral properties of 7-DCCA dye in water/AOT/isooctane reverse micelles.47 We have seen that the absorption peak of 7-DCCA in CD/reverse micelle at w0 = 3 appears at 391 nm and with gradual swelling of the reverse micelle core, the absorption peak undergoes red shift, thereby reaching 399 nm at w0 = 30 as shown in Fig. 1 and Table 1. This absorption peak at w0 = 30 is blue shifted by 7 nm compared to 7-DCCA dye in water/AOT/isooctane reverse micelles. This shows the effect of double confinement of 7-DCCA on the ground state spectral property inside the reverse micelle. For every w0 value the absorption peak of doubly confined dye is blue shifted compared to the singly confined dye in the same reverse micelle. In our present system the absorption peak is red shifted from 391 nm at w0 = 3 to 399 nm at w0 = 30 whereas, the absorption peak underwent red shift from 394 nm at w0 = 3 to 406 nm at w0 = 30 for singly confined dye in the water/AOT/isooctane reverse micelles.47 This clearly indicates that due to double confinement 7-DCCA experiences quite different environment than singly confined 7-DCCA in the same reverse micelles. These absorption peaks however, are blue shifted compared to the absorption peak of 7-DCCA in neat water (Fig. 1). This clearly indicates that 7-DCCA experiences certainly a different atmosphere inside the CD/reverse micelle due to double confinement. We can affirmatively say that the 7-DCCA resides completely inside the water pool of the reverse micelles because of two reasons (a) 7-DCCA is highly soluble in water and (b) being complexed with HP-γ-CD it becomes more soluble in water. So it is expected that 7-DCCA + HP-γ-CD complex will reside completely inside the water pool of the reverse micelle. When we have studied the host guest complexation of 7-DCCA ([7-DCCA] = 3 × 10−6 (M)) with HP-γ-CD we had found that the absorption peak of 7-DCCA underwent blue shift from 409 nm in water to 403 nm in the presence of 7.30 mM HP-γ-CD. So, further blue shift of the absorption peak in case of doubly confined dye clearly demonstrates the changing environment inside the reverse micelle due to greater confinement. 7-DCCA is poorly soluble in isooctane and shows three absorption bands located at 413 nm, 402 nm and 390 nm. Coumarin 343 shows mainly two absorption bands in n-heptane at ∼402 nm due to monomer and ∼428 nm due to aggregated form.52 7-DCCA also shows similar feature corresponding to monomer absorption at 390 nm and aggregated absorption at 413 nm in isooctane. In our case, the absorption band at w0 = 3 appears at 391 nm, so we are expecting the emission from the monomer form of the dye, which is highly soluble in water (Fig. S1†).
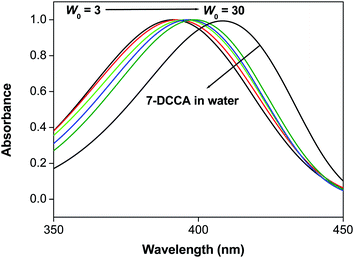 |
| | Fig. 1 The change of the absorption spectral peak position of 7-DCCA in the doubly confined system in the reverse micelles with change in w0 from 0 to 30 and in neat water. | |
Table 1 The photophysical parameters of 7-DCCA in the doubly confined system in the water/AOT/isooctane reverse micelles at different w0 values
| Sr. no. |
System |
w
0
|
λ
max
abs (nm) |
λ
max
emi (nm) |
ϕ
f
|
τ
f (ns) |
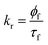
(×108) s−1 |
k
nr (×109) s−1 |
REES (nm) |
| 1. |
7-DCCA doubly confined in the reverse micelles |
3 |
391 |
453 |
0.59 |
2.020 |
2.921 |
0.203 |
6 |
| 2. |
5 |
393 |
457 |
0.39 |
1.503 |
2.595 |
0.406 |
4 |
| 3. |
10 |
395 |
460 |
0.22 |
0.898 |
2.450 |
0.869 |
2 |
| 4. |
20 |
397 |
462 |
0.17 |
0.687 |
2.475 |
1.208 |
1 |
| 5. |
30 |
399 |
462 |
0.15 |
0.632 |
2.373 |
1.345 |
1 |
We have compared the fluorescence emission properties of 7-DCCA in CD/reverse micelles with the emission spectra of 7-DCCA in the reverse micelles. We have found that for all the w0 values the fluorescence emission peak of 7-DCCA in doubly confined system (λexi = 405 nm) is red shifted compared to that of 7-DCCA in singly confined system. The fluorescence emission peak of 7-DCCA in doubly confined system appears at 453 nm at w0 = 3 and with gradual swelling of the reverse micelle core the fluorescence emission peak gets red shifted to 462 nm at w0 = 30 (Fig. 2 and Table 1). In our previous study we have observed that 7-DCCA inside the reverse micellar core shows its emission peak at 450 nm at w0 = 3 and 454 nm when w0 = 30.47 This clearly indicates that for all the w0 values the emission peak of 7-DCCA in doubly confined system inside the core of the reverse micelle is red shifted compared to the singly confined dye inside the reverse micelle. Moreover, 7-DCCA in water solution shows a prominent emission peak at 470 nm. When encapsulated by HP-γ-CD it shows emission band at 461 nm. When this complex is trapped further inside the core of water/AOT/isooctane reverse micelles, we have found that the emission peak undergoes further blue shift to 453 nm at w0 = 3. Probably, this is due to the less polar environment faced by the doubly confined dye inside the core of reverse micelle. We know that the water molecules present inside the core of reverse micelle are less polar compared to the bulk water molecules.53,54 With the gradual addition of water in the reverse micelles the fluorescence emission peak gradually undergoes red shifting as the polarity of the water pool increases. Moreover, when the dye-macrocycle complex is entrapped inside the core of reverse micelle the dye faces more restricted environment inside the core of reverse micelle. This is due to the fact that the viscosity of water inside the core of reverse micelle is higher than the bulk water and this viscosity gradually decreases with the gradual increase of water content.55 So the high viscosity of water inside the reverse micelle core at lower w0 value imposes greater restriction to the doubly confined dye to form TICT state, thereby causing the blue shifted emission peak in the reverse micelle. This high restriction imposed on the 7-DCCA due to double confinement in the core of reverse micelle causes a high quantum yield of the dye (Table 1), compared to single confinement of dye in the same reverse micelle.47 The fluorescence quantum yield of the doubly confined dye gradually decreases from 0.59 at w0 = 3 to 0.15 at w0 = 30, demonstrating that with gradual swelling of the water pool, the dye-macrocycle complex experiences gradually decreasing restriction, thereby increasing the tendency to form TICT state by the rotation of diethylamino group in the excited state (λexi = 405 nm). This shows that the dye experiences greater restriction imposed on it due to double confinement than the single confinement inside the reverse micelle. This difference in fluorescence quantum yield value is prominent upto w0 = 10, between doubly confined dye and the singly confined dye. Beyond w0 = 10 the difference gradually starts to decrease. At w0 = 20, the fluorescence quantum yield is 0.17 for the doubly confined dye, the same is 0.15 for singly confined dye inside the reverse micelle. At w0 = 30 the quantum yield values are almost same for both the cases. 7-DCCA exhibited very low solubility in nonpolar solvent. In isooctane it shows the emission spectrum which is quite similar in shape as that of Coumarin 343 in cyclohexane. Correa and Levinger reported the emission spectra of Coumarin 343 in cyclohexane and reported the emission from both the monomer and aggregated form.56 A similar type of emission feature is observed in the case of 7-DCCA in isooctane. As in the present experimental condition, the dye is complexed with HP-γ-CD and the dye concentration in the reverse micelle is sufficiently low so the aggregate formation is inhibited. Moreover, the emission spectra of the dye–macrocycle complex inside the reverse micelle at w0 = 3 is sufficiently red shifted compared to the emission bands of 7-DCCA in isooctane indicating that the dye–macrocyclic complex residing in the water pool and not in the nonpolar solvent (Fig. S2†). At w0 = 3, the dye-macrocycle complex is expected to reside near the interface and experiences very high restriction, making its fluorescence quantum yield value very high compared to that of singly confined dye in the reverse micelle at same w0.
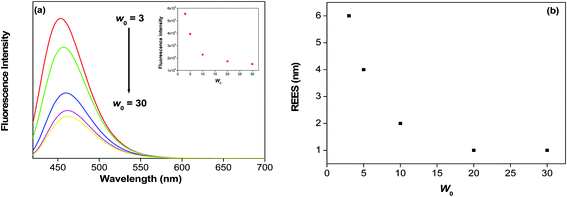 |
| | Fig. 2 (a) The change in steady state fluorescence emission spectra of 7-DCCA in the doubly confined system in reverse micelles with increasing w0 value (λexi = 405 nm). Inset shows the variation of fluorescence intensity with the variation of w0 value. (b) The variation of red edge excitation shift (REES) with varying the w0 value. | |
It is known that the addition of cyclodextrin (HP-γ-CD) and 7-DCCA can cause the perturbation of the reverse micelle and the properties of reverse micelles are not like before.41 We have previously seen that 7-DCCA shows emission spectra at 470 nm in bulk water and when complexed with HP-γ-CD shows a blue shifted emission at 461 nm. Similarly, at w0 = 3 it is observed that doubly confined dye shows a red shifted emission compared to the singly confined dye in the same reverse micelle at w0 = 3. Again this red shift increases with the increase of w0 value and this red shifted emission for the dye-macrocyclic complex is highest at w0 = 30. This red shifted fluorescence emission peak of 7-DCCA in doubly confined system compared to singly confined system in the same reverse micelle at different w0 values are clear evidence of interaction of dye with macrocycle inside the core of reverse micelles. Similar kind of observation was observed by Jane et al. who showed the interaction between a hydrophobic dye Coumarin 522 with comparatively less water soluble β-cyclodextrin.41 They reported three probable types of formation of supramolecular host–guest complex inside the core of the reverse micelle. The first assumption dealt with the formation of unperturbed complex between the dye and macrocycle. Since the water pool at the core of reverse micelle is perturbed after the addition of solutes, this kind of unperturbed host guest complexation in the water pool of reverse micelle is unlikely to take place. A second assumption, which says minimal interaction between the dye and the macrocycle is ruled out, as here both the dye and the macrocycle HP-γ-CD are sufficiently hydrophilic in nature. In our system, the red shifting of emission spectra of the doubly confined dye compared to singly confined dye inside the core of reverse micelles ranges from 3 nm at w0 = 3 to 8 nm at w0 = 30. These further rules out the second assumption of minimal interaction between the dye and the macrocycle inside the water pool of the reverse micelles. The third assumption deals with the formation of the dye-supramolecular complex inside the core of reverse micelle in the perturbed water pool. Here the dye-HP-γ-CD complexes exist in the water pool inside the reverse micelle. In our case, the red shifted emission spectra compared to the emission spectra of singly confined dye inside the reverse micelle at every w0 value provides definite proof of the formation of a supramolecular host–guest complex inside the core of reverse micelle. Now we need the explanation of red shifted emission spectra of dye in the doubly confined complex inside the core of reverse micelle compared to that of single confinement dye inside the same medium. The two simultaneous and complementary processes inside the cavity of reverse micelles can also account for the observed red shift. Jane et al. provided the idea of these two processes; one of which is the trapping of both dye and macrocycle inside the cavity of reverse micelle and second being the perturbation of the reverse micelle by both the dye and macrocycle, thereby increasing the polarity of water trapped in the core of reverse micelles.41 The water molecules trapped inside the cavity of HP-γ-CD may also contribute to the observed red shift. Now perturbation of the reverse micelle by trapping both the dye and the HP-γ-CD causes the greater mobilisation of the water molecules inside the cavity of the cyclodextrin, thereby causing the increase of polarity inside the cavity of HP-γ-CD and red shifting the emission peak compared to that of pure dye inside the water reverse micelle. With the gradual swelling of the reverse micelle the number of free water molecules increases. These are more polar than the bound water molecules and mobilisation of these free water molecules inside the cavity of HP-γ-CD causes the greater red shifting of the emission spectra compared to that of singly confined dye in the same medium. Again, the existence of a supramolecular host–guest complex in the water pool of the reverse micelle is evident from the clear difference of full width at half maximum (FWHM) of dye in doubly and singly confined systems. In our present system the FWHM values decreases in a regular pattern whereas, in case of singly confined dye in the core of reverse micelle FWHM increased in a continuous way. The difference of FWHM values for doubly and the singly confined dye inside the reverse micelle at every w0 value indicates the existence of a dye macrocycle complex in the core of reverse micelle. The lower FWHM for our present system compared to that of single confinement dye in water/AOT/isooctane reverse micelles47 depicts greater confinement of the dye in our present system.
3.2. Time resolved emission spectral studies
We studied the time resolved emission spectra (λexi = 405 nm) in order to have a better understanding about the double confinement of dye inside the reverse micelle. We fitted all the decay profiles by biexponential function. We found that at w0 = 3 the fluorescence emission decay consists of two components, one of which is of 1.313 ns (43%, fast component) and another one being 2.553 ns (57%, slow component). With the gradual increase of water pool size inside the core of reverse micelle we have found that the fast component decreases in timescale but at the same time its weight percentage is gradually increasing. It is found that the timescale value for the fast component gradually decreases from 1.313 ns at w0 = 3 to 0.552 ns at w0 = 30. The weight percentage of this component gradually increases from 43% to 94%. On the other hand, the slow component also gradually decreases in its timescale from 2.553 ns to 1.893 ns. The weight percentage of this component also gradually decreases from 57% at w0 = 3 to 6% at w0 = 30 (Fig. S3† and Table 2). This indicates the gradual relaxing environment around the dye-macrocycle complex with gradual swelling of the water pool. This decrease in restriction around the dye-macrocyclic complex is also reflected from the decrease of average decay time with the gradual increase of w0 value (Fig. 3(a) and Table 1). This gradual decrease in average fluorescence decay time is mainly due to the gradual decrease of restrictedness of the reverse micellar core, causing the increase of the non radiative decay pathway. Now we have found in our system that for w0 = 3 the fluorescence decay time of 7-DCCA is greater for doubly confined complex than the singly confined dye inside the water/AOT/isooctane reverse micelle at the same w0 value. When the water pool is small enough at w0 = 3, the semi frozen nature of the water molecules due to the presence of a predominantly large number of bound water molecules causes the greater restriction and greater confinement of 7-DCCA inside the pool. This causes the greater fluorescence decay time of dye in doubly confined system compared to that in singly confined system in the same medium. With the gradual increase of size of the water pool inside the core of reverse micelle the number of free water molecules increases causing the greater mobilisation and greater polarity of water. This gradually releases the restriction and the dye-macrocycle complex experiences comparatively less confinement. Even at w0 = 30 the fluorescence decay time of 7-DCCA in doubly confined system is higher than that single confined system in water/AOT/isooctane reverse micelle at w0 = 30. This clearly showed that even at very high w0 value, the dye experiences greater restricted environment in doubly confined system compared to single confined system at the same w0 = 30 value. This clearly demonstrates the probability of interaction between the dye 7-DCCA and macrocycle HP-γ-CD inside the core of reverse micelle in the confined water pool. We have found out the partition coefficient of 7-DCCA when complexed with HP-γ-CD in water to know the percentage of complex or free dye present in the solution, when adding water solution of dye-macrocyclic complex in the isooctane–AOT mixture. This will also help us to know the amount of dye formed complex with HP-γ-CD in the water solution used to form the reverse micelles. We have used the following equation for finding out the partition coefficient of dye in between water and the macrocycle HP-γ-CD:57,58| | 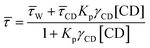 | (10) |
where, ![[small tau, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d4.gif) , W, CD are the fluorescence average decay time, fluorescence decay time in water, fluorescence decay time of 7-DCCA bound to HP-γ-CD. Kp and γCD stand for partition coefficient and the molar volume of HP-γ-CD. From the plot of against [CD] we have found that the partition coefficient is 1.36 × 102 together with γCD value 1.36 dm3 mol−1. The plot gives a very good fitting with the fitting parameter R2 = 0.997 (Fig. S4†). The large partition coefficient value indicates a very strong binding of the dye with the macrocycle HP-γ-CD in water and it is observed that about 99.3% dye is complexed with the macrocycle in water, leaving very small amount of un-complexed dye in water. So most of the dye encapsulated inside the core of reverse micelle as complex with HP-γ-CD. We have previously mentioned that this dye-macrocycle complex is quite different in nature inside the water pool of the reverse micelle than in the bulk water as evident from the fluorescence decay time. Moreover, in bulk water the decay of 7-DCCA complexed with HP-γ-CD is tri-exponential in nature, whereas that inside the core of reverse micelle is completely biexponential in nature. This also clearly demonstrates the difference of nature of complex of 7-DCCA with HP-γ-CD in the water pool of reverse micelle and bulk water. Initially, when the pool is small in size the dye-macrocycle complex mainly resides near the interface. With the gradual increase in size of the water pool it shows tendency to move towards the centre of the pool, because the dye 7-DCCA has been made more hydrophilic by encapsulating it inside the cavity of highly water soluble macrocycle HP-γ-CD, so that no portion of dye resides in the nonpolar phase. This ensures that the dye-macrocycle complex completely reside inside the core of reverse micelle in water pool. With the gradual increase of the pool size (i.e. with increasing w0 value) the dye-macrocycle complex shows greater propensity to move towards the core of the pool from the interfacial region. Since, the complex is more hydrophilic than the uncapsulated dye and in the large reverse micelles the number of free water molecules increases, thereby increasing the polarity of the core of the water pool. For the dye 7-DCCA, TICT formation is the main nonradiative deactivation pathway and this deactivation goes through the formation of ICT state, and it further undergoes ultrafast twisting motion to produce TICT state. The rate constant of radiative and non radiative deactivation pathway is provided by the following equations:
, W, CD are the fluorescence average decay time, fluorescence decay time in water, fluorescence decay time of 7-DCCA bound to HP-γ-CD. Kp and γCD stand for partition coefficient and the molar volume of HP-γ-CD. From the plot of against [CD] we have found that the partition coefficient is 1.36 × 102 together with γCD value 1.36 dm3 mol−1. The plot gives a very good fitting with the fitting parameter R2 = 0.997 (Fig. S4†). The large partition coefficient value indicates a very strong binding of the dye with the macrocycle HP-γ-CD in water and it is observed that about 99.3% dye is complexed with the macrocycle in water, leaving very small amount of un-complexed dye in water. So most of the dye encapsulated inside the core of reverse micelle as complex with HP-γ-CD. We have previously mentioned that this dye-macrocycle complex is quite different in nature inside the water pool of the reverse micelle than in the bulk water as evident from the fluorescence decay time. Moreover, in bulk water the decay of 7-DCCA complexed with HP-γ-CD is tri-exponential in nature, whereas that inside the core of reverse micelle is completely biexponential in nature. This also clearly demonstrates the difference of nature of complex of 7-DCCA with HP-γ-CD in the water pool of reverse micelle and bulk water. Initially, when the pool is small in size the dye-macrocycle complex mainly resides near the interface. With the gradual increase in size of the water pool it shows tendency to move towards the centre of the pool, because the dye 7-DCCA has been made more hydrophilic by encapsulating it inside the cavity of highly water soluble macrocycle HP-γ-CD, so that no portion of dye resides in the nonpolar phase. This ensures that the dye-macrocycle complex completely reside inside the core of reverse micelle in water pool. With the gradual increase of the pool size (i.e. with increasing w0 value) the dye-macrocycle complex shows greater propensity to move towards the core of the pool from the interfacial region. Since, the complex is more hydrophilic than the uncapsulated dye and in the large reverse micelles the number of free water molecules increases, thereby increasing the polarity of the core of the water pool. For the dye 7-DCCA, TICT formation is the main nonradiative deactivation pathway and this deactivation goes through the formation of ICT state, and it further undergoes ultrafast twisting motion to produce TICT state. The rate constant of radiative and non radiative deactivation pathway is provided by the following equations:| | 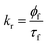 | (11) |
| | 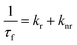 | (12) |
Table 2 The fluorescence lifetime components of 7-DCCA in the doubly confined system in the water/AOT/isooctane reverse micelles at different w0 values
| Sr. no. |
System |
w
0
|
τ
1 (ns) |
a
1
|
τ
2 (ns) |
a
2
|
〈τ〉 (ns) |
χ
2
|
| 1. |
7-DCCA doubly confined in the reverse micelles |
3 |
1.313 |
0.43 |
2.553 |
0.57 |
2.020 |
1.02 |
| 2. |
5 |
1.168 |
0.69 |
2.248 |
0.31 |
1.503 |
1.02 |
| 3. |
10 |
0.782 |
0.90 |
1.943 |
0.10 |
0.898 |
1.01 |
| 4. |
20 |
0.618 |
0.95 |
1.991 |
0.05 |
0.687 |
1.01 |
| 5. |
30 |
0.552 |
0.94 |
1.893 |
0.06 |
0.632 |
1.09 |
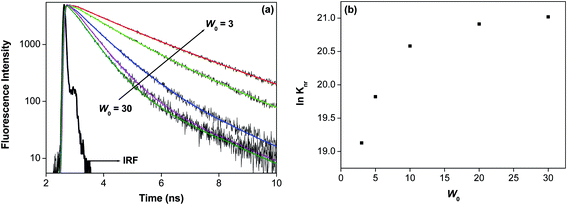 |
| | Fig. 3 (a) The change in fluorescence decay time of 7-DCCA in the doubly confined system in reverse micelles with increase w0 value (λexi = 405 nm). (b) The change of ln(knr) value with increase in w0 value. | |
Comparison of the values of radiative and non radiative decay rate constant, between the doubly and singly confined molecules, helps us to understand the nature of greater confinement of the dye in our present system. The non radiative decay rate constant (knr) in case of singly confined dye is always higher than doubly confined dye. It is observed that in water after addition of 3.41 × 10−4 (M) HP-γ-CD to the aqueous solution of 7-DCCA, the non radiative decay rate constant become 5.68 × 109 s−1. This shows that in double confinement system the restrictedness around the dye is higher compared to both single confinement system and in supramolecular host guest complex in water. This shows that in doubly confined system the greater restrictedness prevents the formation of TICT state so that the nonradiative decay constant becomes significantly lower. We have found that at w0 = 3, the value of knr is 0.203 × 109 s−1 and this constant increases to 1.345 × 109 s−1 at w0 = 30 (Fig. 3(b) and Table 1). This clearly depicts that with the gradual increase of the water pool, the restriction on the dye-macrocycle complex inside the core of the reverse micelle decreases. With the gradual increase of the water pool size the polarity of the water pool increases as polarity of the water pool is directly related to the w0 value.59,60 Moreover, with the gradual swelling of the water pool the viscosity of the pool decreases. So with the gradual rise of size of the water pool the more polar water molecules enter inside the cavity of the cyclodextrin HP-γ-CD causing nonradiative deactivation of the excited state. Moreover, decreasing viscosity of water with the gradual increase of the w0 value increases the nonradiative decay rate of the dye entrapped inside the cavity of the HP-γ-CD. The formation of the dye-macrocycle complex inside the core of reverse micelle is also evident from the value of knr, it is lower for our present system than that of singly confined dye in the same system at all the w0 values.47 This clearly indicates that the dye is undergoing interaction with the macrocycle inside the core water/AOT/isooctane reverse micelles. It is found that for doubly confined system at w0 = 3 the knr value is 0.203 × 109 s−1 whereas, at w0 = 3 for singly confined dye inside the reverse micelle core47knr value is 0.335 × 109 s−1. This shows greater confinement of the dye in the doubly confined system. Even at w0 = 30 the knr value of doubly confined dye is lower than the single confined dye inside the same system. This clearly demonstrates that even at w0 = 30 the interaction between the dye and the macrocycle persists in the water pool.
3.3 Solvation dynamics
We have studied the solvation dynamics in our present system. We found that for our present system, the decay of C(t) was fitted biexponentially upto w0 = 10 and single exponentially beyond w0 = 10 (Fig. 4 and Table 3). The time resolved emission spectra (TRES) was constructed for obtaining an idea about the dynamic Stokes shift or time dependent Stokes shift (TDSS). The time resolved emission spectra (TRES) demonstrating the time dependent Stokes shift (TDSS) at two different w0 values are presented in Fig. S5.† The emission wavelength dependent emission decays ranging from red end to the blue end of the emission peak at two different w0 values are shown in Fig. 5. We have found that the average solvent relaxation time gradually decreases with the gradual increase of size of the water pool (Table 3). This is due to the increase of free water molecules with the gradual increase of water pool size. We have found that at w0 = 3 the solvent relaxation time consists of two components. One of those components is slow and the other one is fast. The time scale of the fast component is 0.190 ns, this contributes 34% to the total solvent relaxation, and the slow component of 1.570 ns contributes 66% of the total solvent relaxation. The most important aspect of this solvent relaxation is that the timescale of the fast component is higher than that in the case of singly confined dye in reverse micelle at w0 = 3, although the weight percentage of the fast component is less than that our previous experiment at same w0 value.47 The timescale of the slow component and the corresponding weight percentage in our present system is higher than the singly confined dye in the reverse micelles. This result is quite obvious from the point of view that the water molecules inside the pool of reverse micelle enter inside the cavity of macrocycle HP-γ-CD. As a result the restricted and immobilised nature of water molecule increases. This causes the increase of the timescale of both the fast and slow components. The weight percentage of the slow component is higher in the present system compared to singly confined system at w0 = 3. This confirm our previous assumption that initially at w0 = 3 (where, the pool size is sufficiently small) the dye macrocycle complex use to reside near the interfacial region. The solvent relaxation time consists of two components upto w0 = 10, after that the solvent relaxation time consists of only one component as like singly confined system. Upto w0 = 5 the average solvent relaxation time is significantly high for our present system compared to that in singly confined system. Here, the slow component is higher in time scale than that in singly confined dye in the same revere micelle, although the weight percentage is slightly lower than that in singly confined dye. The slow component is due to the relaxation of the bound water. This demonstrated that the dye-macrocycle complex is moving towards the core of the reverse micelles with the increase of size of the water pool. This is also supported by the fact that both the bound and free water molecules are in dynamic equilibrium and undergoes continuous exchange with each other. So the dye-macrocycle complexes staying near the interfacial region move towards the core of the pool with the gradual swelling of the reverse micelles. After w0 = 5, the average rotational relaxation time gradually starts to become comparable with singly confined medium. We have previously said that the addition of both the HP-γ-CD and dye causes the perturbation of the water pool. So this perturbation causes the greater mobilisation of the water molecules inside the cavity of HP-γ-CD thereby making the solvent relaxation fast. At w0 = 10 the fast component is higher in timescale than that in singly confined dye in reverse micelle and also the weight percentage is higher. This shows that free water molecules are moving inside the cavity of the cyclodextrin thereby increasing the timescale. Beyond the w0 = 10, the solvent relaxation time consists of only one component and this component is almost same as that in single confined system. This indicates that the supramolecular dye-macrocyclic complex has moved towards the centre of the pool, where only one kind of water molecule undergoes relaxation. However, in our case we have found that the dynamic Stokes shift or TDSS value is lower for every w0 than that in 7-DCCA/water/AOT/isooctane. This also demonstrates the presence of 7-DCCA-HP-γ-CD complex inside the core of reverse micelle and also indicates that even at w0 = 30 this complex persists in the water pool.
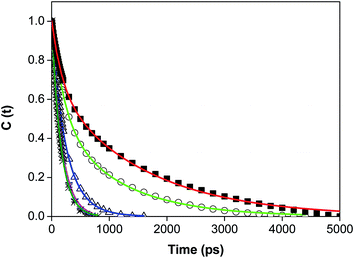 |
| | Fig. 4 The decays of C(t) of 7-DCCA in the doubly confined system in reverse micelles at different w0 (λexi = 405 nm) (i) w0 = 3 (■), (ii) w0 = 5 (○), (iii) w0 = 10 (Δ), (iv) w0 = 20 (☆), (v) w0 = 30 (*). | |
Table 3 The decay characteristics of C(t) of 7-DCCA in the doubly confined system
| Sr. no. |
System |
w
0
|
Δν (cm−1) |
τ
1 (ns) |
a
1
|
τ
2 (ns) |
a
2
|
〈τ〉 (ns) |
ΔG≠ (cal mol−1) |
λ
exi (nm) |
| 1. |
7-DCCA doubly confined in the reverse micelles |
3 |
666 |
0.190 |
0.34 |
1.570 |
0.66 |
1.100 |
5208 |
405 |
| 2. |
5 |
409 |
0.218 |
0.49 |
1.097 |
0.51 |
0.666 |
4913 |
| 3. |
10 |
395 |
0.183 |
0.49 |
0.320 |
0.51 |
0.253 |
4342 |
| 4. |
20 |
431 |
— |
— |
0.173 |
1 |
0.173 |
4117 |
| 5. |
30 |
261 |
— |
— |
0.159 |
1 |
0.159 |
4068 |
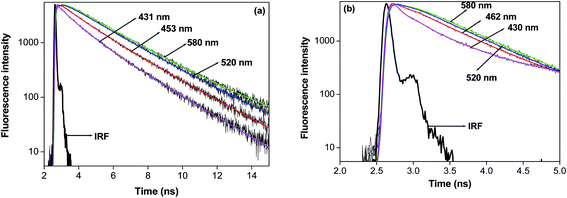 |
| | Fig. 5 The wavelength dependent fluorescence decays of 7-DCCA in the doubly confined system in reverse micelles at (a) w0 = 3 and (b) w0 = 20. | |
Here in order to quantify the energetic associated with the solvation process we have found out the values of ΔG≠ using the Eyring equation:61,62
| | 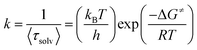 | (13) |
where Δ
G≠ is the free energy change of the solvation process,
kB and
h are the Boltzmann constant and Plank's constant respectively. Here
k stand for rate constant for solvent relaxation process. Using this equation we have found that the free energy change of the solvation process gradually changes from 5.21 kcal mol
−1 at
w0 = 3 to 4.07 kcal mol
−1 at
w0 = 30 (
Table 3). Here Δ
G≠ = (Δ
G* + Δ
G0), where Δ
G* and Δ
G0 stand for the activation energy of desorption from the surface of the biomolecule and the excess hydrogen bond energy of water with the biomolecule (over that in the bulk). According to the model proposed by Nandi and Bagchi we have taken the value of Δ
G* to be constant (1.5
kBT). As the Δ
G* value is very small, the contribution of Δ
G* in Δ
G≠ can be neglected. Then the value of Δ
G≠ is almost equal to Δ
G0. Pal
et al. had studied the femtosecond dynamics of bulk water and calculated the Gibbs free energy of solvation. They found the free energy to be ∼750 cal mol
−1.
63 However, in our present system we have found that the Δ
G≠ value ranges from 5.21 kcal mol
−1 at
w0 = 3 and 4.07 kcal mol
−1 at
w0 = 30. These are much larger than that in water clearly depicting the rupture of hydrogen bonded network of water inside the water pool of reverse micelle. We had found that for all the
w0 values especially for 3 and 5 there are clear differences of Δ
G≠ values of our present system with single confined system. After
w0 = 5 for both the systems Δ
G≠ values become almost same. This further confirms our previous assumption that the addition of both HP-γ-CD and 7-DCCA perturbs the water pool of the reverse micelle and this perturbation causes the difference in average solvent relaxation times in our present system and with that of single confined system.
3.4 Red edge excitation shift
We reported the red edge excitation shift (REES) of 7-DCCA inside the core of water/AOT/isooctane reverse micelles at different w0 values.47 In our present system, we have also found REES. We have found that at w0 = 3, with changing the excitation wavelength from 405 nm to 435 nm, the emission peak shifts by 6 nm. With the gradual increase of the size of the water pool of reverse micelle the REES value gradually decreases as shown in Table 1 and Fig. 2(b). At w0 = 20 with changing λexi from 405 nm to 435 nm the REES value is only 1 nm, whereas for singly confined system the observed REES is 5 nm for changing the λexi from 405 nm to 425 nm. This indicates a different type of environment faced by the dye inside the core of reverse micelle at even w0 = 20 in the doubly confined system. This also shows that the dye is buried inside the cavity of HP-γ-CD in the core of reverse micelle. This causes the significantly less REES in our present system compared to singly confined system. In singly confined system, we have found that the extent of REES gradually decreases with gradual swelling of the water pool inside the core of reverse micelle. REES is a technique; it provides valuable information about the relative rate of solvent relaxation. Moreover, this technique helps us to directly monitor the microenvironment and dynamics around the fluorophore in different restricted and microheterogeneous medium. There have been reports of observing REES in water reverse micelle by different groups.56,64–67 REES is prominently observed for a number of fluorophores in different media like frozen media, in different vitrified solution, highly viscous solution and polymer matrices. In our present study, we have seen that, the solvent relaxation time is smaller than the fluorescence decay time (τf) of the fluorophore at every w0 value. This may be due to the specific solute–solvent interaction between the doubly confined dye and water molecule inside the core of reverse micelle. This type of hydrogen bonding interaction is quite common phenomenon for 7-DCCA with polar protic solvent molecules. This substantially modulates the photophysical behaviour of 7-DCCA.48 So this specific solute solvent interaction is mainly responsible for the observed REES in our present system. The water molecules trapped inside the core of reverse micelle act as hydrogen bond donor, whereas 7-DCCA acts as hydrogen bond acceptor. Samanta and co-worker reported similar kind of phenomenon of specific solute solvent interaction between the dye HNBD and RTIL, where the RTIL acts as hydrogen bond acceptor facilitating the photoselection.68 This causes the REES of HNBD in RTIL. With the gradual increase of size of the water pool the extent of red edge excitation shift gradually decreases. This is mainly due to the fact that, with the gradual increase of size of the water pool, fluidity of the medium increases. This causes the faster relaxation of the confined water molecules inside the core of reverse micelles thereby decreasing the extent of REES. So this supports our assumption that with the gradual increase of hydration, the extent of REES gradually decreases, implying that the motional restriction is gradually decreasing with the increase in water content.
3.5 Time resolved anisotropy studies
Rotational relaxation studies further helps us to understand the formation of a supramolecular host–guest complex inside the core of reverse micelle. In pure water 7-DCCA shows rotational relaxation time of ∼90 ps. In water solution of 7-DCCA (3 × 10−6 (M)) after the addition of 7.30 mM HP-γ-CD the rotational relaxation time increases to 0.684 ns. This indicates the formation of a supramolecular host guest complex between the dye 7-DCCA and macrocycle HP-γ-CD. Now after the incorporation of 7-DCCA-HP-γ-CD host guest complex inside the core of reverse micelle, the average rotational relaxation time increases significantly to 3.561 ns at w0 = 3. The rotational relaxation time gradually decreases with the gradual increase of w0 value i.e. the size of the water pool inside the core of reverse micelle (Fig. 6 and Table 4). At w0 = 30 the average rotational relaxation time becomes 0.780 ns. The most noteworthy thing of this study is that in the case of doubly confined system at every w0 value the rotational relaxation time is prominently higher than that in singly confined system. We have fitted the anisotropy decays by biexponential function. We have found that for all the w0 values, both the components are higher in timescale than that in singly confined system.47 This clearly indicates that after the double confinement of the dye inside the core of water/AOT/isooctane reverse micelle the rotational relaxation of the 7-DCCA decreases. This indicates the existence of the dye-macrocycle complex inside the core of reverse micelle. We have observed that even at w0 = 30 the average rotational relaxation time of 7-DCCA in the doubly confined system is higher than that in single confined system. This proof the existence of supramolecular host guest complexation inside the core of reverse micelle and this complex exist even at w0 = 30. With the gradual increase of size of the water pool, the restriction over the dye-macrocycle complex gradually decreases, so the rotational relaxation time also gradually decreases. When the size of reverse micelle is small, the frozen nature of the water pool prevents the rotational motion of the dye macrocycle complex, causing high rotational relaxation time. This relaxation time is higher than that of singly confined dye at same w0 value. With the gradual swelling of the reverse micelle the movement of the water molecules inside the core increases and it decreases the restriction on the rotation of the dye-macrocycle complex, hence reducing the rotational relaxation time.
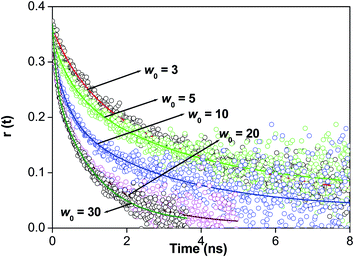 |
| | Fig. 6 The overlay of time resolved anisotropy decays of 7-DCCA in the doubly confined system at different w0 values. | |
Table 4 The rotational relaxation time of 7-DCCA in the doubly confined system
| Sr. no. |
System |
w
0
|
λ
exi (nm) |
r
0
|
τ
1 (ns) |
(1 − β) |
τ
2 (ns) |
β
|
〈τ〉 (ns) |
χ
2
|
| 1. |
7-DCCA doubly confined in the reverse micelles |
3 |
405 |
0.362 |
0.840 |
0.29 |
4.673 |
0.71 |
3.561 |
1.128 |
| 2. |
5 |
0.347 |
0.485 |
0.30 |
3.290 |
0.70 |
2.450 |
1.044 |
| 3. |
10 |
0.375 |
0.392 |
0.42 |
2.863 |
0.58 |
1.825 |
1.072 |
| 4. |
20 |
0.339 |
0.196 |
0.30 |
1.365 |
0.70 |
1.014 |
1.142 |
| 5. |
30 |
0.385 |
0.100 |
0.32 |
1.100 |
0.68 |
0.780 |
1.189 |
4. Conclusion
In this study we have shown the double confinement of a hydrophilic dye 7-DCCA in CD/reverse micelle system, by first encapsulating dye in HP-γ-CD and then trapping the complex inside the core of the reverse micelle. The absorption spectra of 7-DCCA in doubly confined system shows significant blue shift compared to the singly confined system in the reverse micelle. We have observed red shifted emission spectra of 7-DCCA in our present system at every w0 value than the singly confined system. The fluorescence decay times showed the effect of double confinement on the photophysics of 7-DCCA in the reverse micelles. Solvent relaxation dynamics shows that the dye 7-DCCA faces greater restriction in the doubly confined system than in the singly confined system in the reverse micelle. This type of double confinement in the presence of supramolecular host in the water pool of reverse micelle is further substantiated by the time resolved anisotropy study. The rotational relaxation time of 7-DCCA in the doubly confined system is always higher than the singly confined system. This clearly indicates that due to the double confinement of 7-DCCA inside the reverse micelle the rotational relaxation time is higher compared to the single confined system.
Acknowledgements
D. S. is thankful to IIT Patna, India for research facilities. A. C. is thankful to CSIR, New Delhi, for research fellowship. B. M. is thankful to IIT Patna, for research fellowship.
References
- P. Verma and H. Pal, J. Phys. Chem. A, 2013, 117, 12409 CrossRef CAS PubMed.
- M. Porel, A. Klimczak, M. Freitag, E. Galoppini and V. Ramamurthy, Langmuir, 2012, 28, 3355 CrossRef CAS PubMed.
- S. Bairu and G. Ramakrishna, J. Phys. Chem. B, 2013, 117, 10484 CrossRef CAS PubMed.
- E. Krystkowiak, K. Dobek and A. Maciejewskia, Photochem. Photobiol. Sci., 2013, 12, 446 CAS.
- E. Krystkowiak and A. Maciejewski, Phys. Chem. Chem. Phys., 2011, 13, 11317 RSC.
- P. Zhou, P. Song, J. Liu, K. Han and G. He, Phys. Chem. Chem. Phys., 2009, 11, 9440 RSC.
- D. Roy, S. Pionteka and R. A. Walker, Phys. Chem. Chem. Phys., 2011, 13, 14758 RSC.
- A. Nag and K. Bhattacharyya, Chem. Phys. Lett., 1990, 169, 12 CrossRef CAS.
- B. B. Raju and T. S. Varadarajan, J. Phys. Chem., 1994, 98, 8903 CrossRef.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899 CrossRef PubMed.
- A. S. Satpati, M. Kumbhakar, S. Nath and H. Pal, Photochem. Photobiol., 2009, 85, 119 CrossRef CAS PubMed.
- W. Jarzqba, G. C. Walker, A. E. Johnson, M. A. Kahlow and P. F. Barbara, J. Phys. Chem., 1988, 92, 7039 CrossRef.
- S. Vajda, R. Jimenez, S. J. Rosenthal, V. Fidlert, G. R. Fleming and E. W. Castner Jr, J. Chem. Soc., Faraday Trans., 1995, 91, 867 RSC.
- M. A. Kahlow, T. J. Kang and P. F. Barbara, J. Chem. Phys., 1988, 88, 2372 CrossRef CAS PubMed.
-
Structure and Reactivity in Reverse Micelles, ed. M. P. Pileni, Elsevier, Amsterdam, 1981 Search PubMed.
-
Reverse Micelles, ed. P. L. Luisi and B. E. Straube, Plenum Press, New York, 1984 Search PubMed.
- J. J. Silber, A. Biasutti, E. Abuinb and E. Lissi, Adv. Colloid Interface Sci., 1999, 82, 189 CrossRef CAS.
- T. K. De and A. Maitra, Adv. Colloid Interface Sci., 1995, 59, 95 CrossRef CAS.
- B. Halle, Philos. Trans. R. Soc. London, Ser. B, 2004, 359, 1207 CrossRef CAS PubMed.
- B. Bagchi, Chem. Phys. Lett., 2012, 529, 1 CrossRef CAS PubMed.
- K. Bhattacharyya, Chem. Commun., 2008, 2848 RSC.
- S. K. Pal and A. H. Zewail, Chem. Rev., 2004, 104, 2099 CrossRef CAS PubMed.
- S. P. Moulika and B. K. Paul, Adv. Colloid Interface Sci., 1998, 78, 99 CrossRef.
- H. Hauser, G. Haering, A. Pande and P. L. Luisi, J. Phys. Chem., 1989, 93, 7869 CrossRef CAS.
- M. Wong, J. K. Thomas and M. Gratzel, J. Am. Chem. Soc., 1976, 98, 2391 CrossRef CAS.
- K. K. Karukstis, A. A. Frazier, D. S. Martula and J. A. Whiles, J. Phys. Chem., 1996, 100, 11133 CrossRef CAS.
- A. Amararene, M. Gindre, J.-Y. L. Huérou, C. Nicot, W. Urbach and M. Waks, J. Phys. Chem. B, 1997, 101, 10751 CrossRef CAS.
- I. R. Piletic, D. E. Moilanen, D. B. Spry, N. E. Levinger and M. D. Fayer, J. Phys. Chem. A, 2006, 110, 4985 CrossRef CAS PubMed.
- D. E. Moilanen, N. E. Levinger, D. B. Spry and M. D. Fayer, J. Am. Chem. Soc., 2007, 129, 14311 CrossRef CAS PubMed.
- J. Chowdhary and B. M. Ladanyi, J. Phys. Chem. A, 2011, 115, 6306 CrossRef CAS PubMed.
- P. A. Pieniazek, Y. S. Lin, J. Chowdhary, B. M. Ladanyi and J. L. Skinner, J. Phys. Chem. B, 2009, 113, 15017 CrossRef CAS PubMed.
- N. Gorski and Y. M. Ostanevich, J. Phys. IV, 1993, 03, 149 CrossRef CAS.
- N. Gorski and Y. M. Ostanevich, Ber. Bunsenges. Phys. Chem., 1990, 94, 737 CrossRef CAS.
- T. K. Jain, M. Varshney and A. Maitra, J. Phys. Chem., 1989, 93, 1409 CrossRef.
- N. Sarkar, K. Das, A. Datta, S. Das and K. Bhattacharyya, J. Phys. Chem., 1996, 100, 10523 CrossRef CAS.
- N. E. Levinger and L. A. Swafford, Annu. Rev. Phys. Chem., 2009, 60, 385 CrossRef CAS PubMed.
- A. Patra, T. Q. Luong, R. K. Mitra and M. Havenith, Phys. Chem. Chem. Phys., 2013, 15, 930 RSC.
- B. B. Raju and S. M. B. Costa, J. Phys. Chem. B, 1999, 103, 4309 CrossRef CAS.
- D. Pant and N. E. Levinger, Langmuir, 2000, 16, 10123 CrossRef CAS.
- D. M. Willard and N. E. Levinger, J. Phys. Chem. B, 2000, 104, 11075 CrossRef CAS.
- E. Jane, V. Szçcs, O. Grancicova, T. Palszegi, M. Zitnan, I. Bugar, D. Lorenc and D. Velic, ChemPhysChem, 2012, 13, 4207 CrossRef CAS PubMed.
- S. H. D. Hassonville, B. Perly, G. Piel, T. V. Hees, V. Barillaro, P. Bertholet and L. Delattre, J. Inclusion Phenom. Macrocyclic Chem., 2002, 44, 289 CrossRef.
- G. Ramakrishna and H. N. Ghosh, J. Phys. Chem. A, 2002, 106, 2545 CrossRef CAS.
- H. Zhang, T. Yu, Y. Zhao, D. Fan, L. Chen, Y. Qiu, L. Qian, K. Zhang and C. Yang, Spectrochim. Acta, Part A, 2008, 69, 1136 CrossRef PubMed.
- A. Chatterjee and D. Seth, Photochem. Photobiol., 2013, 89, 280 CrossRef CAS PubMed.
- C. Tablet, I. Matei, E. Pincu, V. Meltzer and M. Hillebrand, J. Mol. Liq., 2012, 168, 47 CrossRef CAS PubMed.
- A. Chatterjee, B. Maity and D. Seth, Phys. Chem. Chem. Phys., 2013, 15, 1894 RSC.
- G. Jones II, W. R. Jackson, C. Y. Choi and W. R. Bergmark, J. Phys. Chem., 1985, 89, 294 CrossRef.
- G. B. Dutt, J. Phys. Chem. B, 2004, 108, 805 CrossRef CAS.
- M. Maroncelli and G. R. Fleming, J. Chem. Phys., 1987, 86, 6221 CrossRef CAS PubMed.
- J. E. Löfroth, J. Phys. Chem., 1986, 90, 1160 CrossRef.
- A. Sengupta, R. V. Khade and P. Hazra, J. Phys. Chem. A, 2011, 115, 10398 CrossRef CAS PubMed.
- C. G. Blanco, L. J. Rodríguez and M. M. Velázquez, J. Colloid Interface Sci., 1999, 211, 380 CrossRef PubMed.
- D. Sahoo and S. Chakravorti, J. Photochem. Photobiol., A, 2009, 205, 129 CrossRef CAS PubMed.
- M. Hasegawa, T. Sugimura, Y. Shindo and A. Kitahara, Colloids Surf., A, 1996, 109, 305 CrossRef CAS.
- N. M. Correa and N. E. Levinger, J. Phys. Chem. B, 2006, 110, 13050 CrossRef CAS PubMed.
- R. Thakur, A. Das and A. Chakraborty, Phys. Chem. Chem. Phys., 2012, 14, 15369 RSC.
- N. C. Santos, M. Prieto and M. A. R. B. Castanho, Biochemistry, 1998, 37, 8674 CrossRef CAS PubMed.
- S. Senapati and A. Chandra, J. Phys. Chem. B, 2001, 105, 5106 CrossRef CAS.
- M. Belletste, M. Lachapelle and G. Durocher, J. Phys. Chem., 1990, 94, 5337 CrossRef.
- N. Nandi and B. Bagchi, J. Phys. Chem. B, 1997, 101, 10954 CrossRef CAS.
- S. Rakshit, R. Saha, A. Chakraborty and S. K. Pal, Langmuir, 2013, 29, 1808 CrossRef CAS PubMed.
- D. Banerjee, P. K. Verma and S. K. Pal, Photochem. Photobiol. Sci., 2009, 8, 1441 CAS.
- A. Chattopadhyay, Chem. Phys. Lipids, 2003, 122, 3 CrossRef CAS.
- A. M. Durantini, R. D. Falcone, J. J. Silber and N. M. Correa, J. Phys. Chem. B, 2013, 117, 3818 CrossRef CAS PubMed.
- F. Moyano, S. S. Quintana, R. D. Falcone, J. J. Silber and N. M. Correa, J. Phys. Chem. B, 2009, 113, 4284 CrossRef CAS PubMed.
- S. S. Quintana, F. Moyano, R. D. Falcone, J. J. Silber and N. M. Correa, J. Phys. Chem. B, 2009, 113, 6718 CrossRef CAS PubMed.
- A. Samanta, J. Phys. Chem. B, 2006, 110, 13704 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra00965g |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a