
Design, synthesis and anticancer activity of benzofuran derivatives targeting VEGFR-2 tyrosine kinase
Abstract
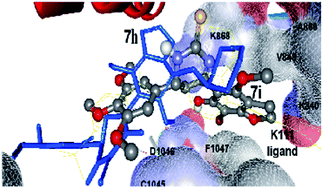
Two series of chalcone and thiopyrimidine benzofuran derivatives were designed, synthesized and evaluated in vitro for their vascular endothelial growth factor receptor (VEGFR-2) inhibitory activity, their cytotoxicity on seventeen human cancer cell lines and their in vivo antiprostate cancer activity. The highest anti-VEGFR-2 activity was demonstrated by 1-(6-hydroxy-4-methoxybenzofuran-5-yl)-3-(4-nitrophenyl)prop-2-en-1-one (6d) exhibiting an IC50 value (1.00 × 10−3 μM) higher than the reference drug Sorafenib (IC50 = 2.00 × 10−3 μM). On the other hand, most of the synthesized compounds showed potent cytotoxicity against most of the tested cell lines and were more potent than the reference drugs, in particular, bromovisnagin (4) exhibited the best activity on the majority of the cell lines with IC50 values ranging from 3.67 × 10−13 to 7.65 × 10−7 μM. Moreover, the synthesized compounds showed significant in vivo antiprostate cancer activity. The docking experiments were performed using the GOLD program on (VEGFR-2) kinase which introduced new information about the enzyme–inhibitor interaction and the potential therapeutic application of the benzofuran scaffold.
 Please wait while we load your content...
Please wait while we load your content...

