Green synthesis of a silver nanoparticle–graphene oxide composite and its application for As(III) detection†
Riyaz Ahmad Dara,
Ninad G. Kharea,
Daniel P. Coleb,
Shashi P. Karnac and
Ashwini Kumar Srivastava*a
aDepartment of Chemistry, University of Mumbai, Vidyanagari, Mumbai-400098, India. E-mail: aksrivastava@chem.mu.ac.in; Tel: +91-22-26543570
bUS Army Research Laboratory, Vehicle Technology Directorate, RDRL-VTM, Aberdeen Proving Ground, Maryland-21005-5069, USA
cUS Army Research Laboratory, Weapons and Materials Research laboratory, RDRL-WM, Aberdeen Proving Ground, Maryland-21005-5069, USA
First published on 11th March 2014
Abstract
We report a facile and green synthetic approach to synthesize a silver nanoparticle (AgNPs)–graphene oxide (GO) composite using beta cyclodextrin as a stabilizing agent and ascorbic acid as a reducing agent. Further, we demonstrate its application as a highly sensitive and selective electrochemical sensor for selective determination of As(III) in the presence of other elements, such as Cu and some organic and inorganic molecules. The GO sheets provided the surface for the reduction of silver ions. The composite can be easily used for the construction of a disposable electrochemical sensor on a glassy carbon electrode (GCE) using a drop deposition method. The composite was characterized by scanning and transmission electron microscopies, energy dispersive X-ray spectroscopy, X-ray diffraction and electrochemical impedance spectroscopy. Cyclic voltammetry and anodic stripping voltammetry measurements were employed to evaluate the electrochemical properties of beta cyclodextrin stabilized AgNPs–GO/GCE towards arsenic(III) detection. The AgNPs–GO film exhibited distinctly higher activity for the anodic stripping analysis of As(III) compared to the GO film alone with approximately three times enhancement of the peak current. This nanostructured electrode applied for As(III) analysis displayed a wide linear range (13.33–375.19 nM), a high sensitivity (180.5(μA μM−1)) including a 0.24 nM detection limit. We demonstrate the real-life application of the developed sensor by selectively determining the As content in ground and river water samples.
Introduction
Nanomaterials of different shapes, sizes, and compositions, have found broad applications in analytical methods. Among these materials, metallic nanoparticles are of immense interest due to their unique electronic, optical, electrical and electrochemical properties with potential applications in a wide range of technologies including chemical and biochemical sensors.1–4 Metal nanoparticle-modified electrodes show dramatically enhanced electrochemical sensitivity due to their large specific surface area and high surface free energy. Some metallic oxides and metallic compounds, such as cobalt oxide, gold and platinum nanoparticles have already been reported to be suitable for electrode modifications.2,5–8 However, such electrodes are costly, display low efficiency and suffer from interference of other metal ions which hinder their commercial applications.2,9–12Among the various metal nanoparticles, silver nanoparticles (AgNPs) offer themselves as a desirable substrate for the preparation of chemically modified electrodes for electrochemical sensing due to high quantum characteristics of small granule diameter, large specific surface area, and the ability for fast electron transfer.13
Literature reports a number of applications of carbon based materials in a variety of fields.14–16 A range of nano scale carbon-based building blocks, including nanotubes, graphene and graphene oxide have attracted significant interest due to their extra ordinary physical and chemical properties, viz., high surface area, high electrical conductivity, strong mechanical strength, biocompatibility and low manufacturing cost.17–19 Graphene oxide (GO), the oxidized form of graphene, bears two-dimensional plane and a large number of oxygen-containing functional groups with disorder on the basal planes and edges.20,21 In addition to retaining some of the desirable functional properties of graphene, it also possesses excellent dispersibility and film-forming features. The covalent oxygen functional groups in GO give rise to remarkable mechanical strength along with molecular-level chemical sensing capability.22,23 Thus, GO is exceptionally suited for use in chemical and biological sensors owing to the nature of the atoms on its surface, its scalable solution-based preparation and a controllable surface defect density that modulates the sensor sensitivity and specificity.24 Recently, covalent or non covalent strategies have been attempted to disperse GO in water through bonding with various types of molecules, such as calixarene,25,26 DNA27 and cyclodextrin.28 At the same time β-cyclodextrin/graphene hybrid nanosheets have been extensively applied as electrochemical sensors.29,30
Composites based on nanosized inorganic particles and clusters represent an attractive class of materials which offer the possibility of tailoring and optimizing their properties for various applications.30–32 GO nanosheets have already been used as supports to disperse and stabilize numerous nanoparticles.30–34 This is possible due to the surface functional groups (–OH, C–O–C, and –COOH) on GO that provide an abundance of reactive sites for the nucleation and binding of metal nanoparticles.35 Nanoparticles are also attractive for contaminant sensing due to their large specific surface area and excellent catalytic properties. Sensors based on nanoparticles can be used for sensing organic, inorganic or biological contaminants.36
In this paper we present a low-cost facile approach for synthesizing AgNPs–GO composite using green chemistry and demonstrate its application as electrochemical sensor for detecting trace-levels of As in the presence of other organic and inorganic contaminants.
Arsenic contamination of ground water is a world-wide serious problem encountered in several countries as it can affect the safety of drinking water.37 As(III) is thought to be more toxic than As(V), due to its reactions with enzymes in human metabolism.38 This causes many diseases such as skin lesions, keratosis, lung cancer, bladder cancer, etc.39–41 The U.S. Environmental Protection Agency (USEPA) established a new arsenic standard in drinking water at 10 ppb in January 2011.42,43 Therefore, the availability of a low-cost and environmental friendly technique for detecting arsenic at or below the designate safety level42,43 in ground water samples is of great importance for preventive/remediation measures against As contamination.
Some non-electrochemical techniques commonly used to determine arsenic are inductively coupled plasma mass spectrometry (ICP-MS),44 electro-spray MS (ES-MS) coupled to chromatography (high performance liquid chromatography and gas chromatography),45 and atomic absorption/fluorescence spectroscopy (AAS/AFS). However, these techniques are expensive and require sample storage and transport, which presents additional challenges in the speciation study. Electrochemical techniques have great advantages for on-site environmental monitoring due to their favourable portability, suitability for automation, short analysis time, low power consumption, and inexpensive equipment. In the past, electrochemical potentiometric stripping techniques have been used for the determination of antimony using a carbon paste electrode employing crown ether and rice husk,46 and bismuth using a carbon paste electrode modified with crown ether and carbon nanotubes.47 To the best of our knowledge, no report has been available regarding the use of a composite of cyclodextrin stabilized AgNPs and graphene oxide as an electrochemical sensor for arsenic detection.
In the present work, the AgNPs–GO composite was expediently synthesized by the reduction of silver ions on GO surface using ascorbic acid as a reducing agent and beta-cyclodextrin as a stabilizing agent. The synthesis was carried out in an aqueous solution, which is a versatile and environmentally-friendly process. The resultants could be easily dispersed into water with common organic solvents to form a stable suspension without any additional protection by polymeric or surfactant stabilizers. The AgNPs–GO composite was then used to fabricate a novel electrochemical sensor to detect and quantify an environmentally toxic element, As. The sensor could provide a larger electrochemically active surface area for the adsorption of arsenic and effectively accelerate the electron transfer between electrode and solution, which could lead to a more rapid and sensitive current response.
Experimental
Materials
Graphite powder (320 mesh, spectrum pure) and paraffin oil, AR grade H2SO4, KMnO4, and H2O2 (30 wt%) were purchased from S. D. Fine Chemicals, India and used as received for the synthesis of GO. AgNO3, ascorbic acid and β-cyclodextrin were purchased from Alfa Aesar Chemical Company. As(III) stock solution (8 × 10−5 M) was prepared with As2O3 dissolved in a small amount of alkali and diluted with distilled water. Doubly-distilled water was used throughout, while 0.1 M H2SO4 was used as the supporting electrolyte.Apparatus
Scanning electron microscopy (SEM) images were obtained from S-4800 field emission SEM system (FEI Quanta 200) operating at 20.0 kV equipped to perform elemental chemical analysis by energy dispersive X-ray spectroscopy (EDX). Transmission electron microscopy (TEM) micrographs were performed using a field emission transmission electron microscope (JEOL JEM-2100F TEM/STEM, Oxford Instruments, USA) operated at 200 kV. X-ray diffraction (XRD) studies were carried out using Maxima 7000S XRD (Shimadzu, Japan).All voltammetric and chrono experiments were performed with a CHI electrochemical workstation (CH Instruments Model CHI1100B series). The conventional three-electrode geometry was utilized. The working electrode was a bare or modified glassy carbon electrode (GCE, 3 mm in diameter), and the auxiliary and reference electrodes were platinum wire and Ag/AgCl, respectively. Electrochemical impedance measurements were performed in 0.1 M KCl containing 1.0 mM Fe(CN)6−3 and Fe(CN)6−4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture) using Eco Chemie, Electrochemical Work Station, model Autolab PGSTAT 30 using Frequency Response Analyser, software version 2.0, measured over a frequency range of 10−1–106 Hz. An Ag/AgCl, a 3 M KCl and a platinum electrode were used as reference and counter electrodes, respectively. The pH measurements were performed using an ELICO LI 120 pH meter.
:1 mixture) using Eco Chemie, Electrochemical Work Station, model Autolab PGSTAT 30 using Frequency Response Analyser, software version 2.0, measured over a frequency range of 10−1–106 Hz. An Ag/AgCl, a 3 M KCl and a platinum electrode were used as reference and counter electrodes, respectively. The pH measurements were performed using an ELICO LI 120 pH meter.
Synthesis of AgNPs–GO composite
GO was synthesized from natural graphite powder by a modified Hummer's method following Hirata et al.48 Generally noble metal nanoparticles are easily obtained using the strong reducing agents such as hydrazine and sodium boro-hydride; however, these reducing agents are toxic and harmful to the environment. In addition, hydrazine and sodium boro-hydride can also reduce the oxygen containing functional groups of GO, resulting in the decrease of sensing performance of the prepared sensor. To avoid such detrimental effects, we used a green and inexpensive chemical synthesis approach. The AgNPs–GO composite was prepared by reducing silver ions directly on GO with ascorbic acid as the reducing agent and β-cyclodextrin as the stabilizing agent. The procedure for the synthesis of silver nanoparticles alone has been discussed earlier.48 In the present typical procedure for the composite synthesis, first, the GO powder (15.0 mg) was dispersed in water (15.0 ml) by sonication for 1 h to form a stable GO colloid. Next, 0.2 g β-cyclodextrin was added to 8.3 ml of 5% AgNO3 and stirred. This solution was added to GO colloid while stirring. Then 10 ml of 2.6% ascorbic acid was added and a precipitate was formed. The precipitate was washed with water repeatedly to remove any impurities. Finally, the obtained product was dried overnight in an oven at 60 °C, resulting in the final AgNPs–GO composite.Preparation of the AgNPs–GO/GCE
First, GCEs were mechanically polished with a 0.05 mm alumina slurry and then sequentially sonicated in dilute nitric acid, anhydrous ethanol and redistilled water for 15 min. Next, the cleaned GCEs were dried under nitrogen stream. 5.0 mg of AgNPs–GO nano-hybrids were added to 2.5 ml dimethylformamide which was sonicated in an ultrasound bath for 30 min to form a stable suspension. The mixed suspension of 10.0 μl was cast onto the GCE surface by a micropipette, and then the suspension was thoroughly dried out under an infrared lamp. Subsequently, the electrode was rinsed by distilled water several times and further dried in air before use; the final obtained electrode is denoted as AgNPs–GO/GCE. For comparison, the GO/GCE was also prepared using the GO dispersion alone and the same procedure as above.Results and discussion
Characterizations of AgNPs–GO composite
The AgNPs–GO composite was characterized by the scanning and transmission electron microscopies (SEM and TEM), energy dispersive X-ray spectroscopy (EDX) and X-ray diffraction (XRD) studies. The SEM and TEM images of the GO and AgNPs–GO composite are shown in Fig. 1. The SEM and TEM images also show the typical crumpled and wrinkled surface of graphene oxide stacked together to form a typical multi-layer structure which provided a large rough surface for further modification. AgNPs were completely distributed on GO sheets (Fig. 1B) with nanoparticles scattered out of the supports, indicating a strong interaction between the GO support and the AgNPs. Fig. 1C and D show the typical TEM images of the synthesized GO and AgNPs–GO composite. A large number of well-dispersed AgNPs are attached onto the surface of GO nanosheets. However, the size of these nanoparticles is between 10–60 nm, and the dispersion is heterogeneous. The reason might be attributed to the stirring inhomogeneity while the formation of AgNPs on GO nanosheets. Some AgNPs were slightly aggregated due to the loading degree close to saturation. Because of GO and silver ions with opposite charges, AgNPs can interact with the GO sheets through physisorption, electrostatic binding or through charge transfer interactions. Highly dispersed AgNPs on the supports with large surface area was expected to improve the catalytic activity and sensor sensitivity. | ||
| Fig. 1 SEM and TEM images of GO film (A and C) and AgNPs–GO composite film (B and D). | ||
The composite formation was further characterized by EDX. The corresponding EDX spectra of GO and GO–AgNPs (Fig. 2A and B) show peaks corresponding to C, O and Ag elements, confirming the existence of metallic AgNPs on the surface of the GO nanosheets. The presence of O indicates the oxygen-containing groups generated during GO synthesis, which remain unreduced during the AgNPs–GO composite synthesis. The atomic and weight ratios of O/Ag in nanocomposite were 1/10.5 and 1/44.1, respectively, indicating the loading of AgNPs on the GO surface, as observed in the SEM images. This confirms that the AgNPs were successfully deposited on the GO. The XRD profile of AgNPs in the composite (Fig. 2Ca) showed peaks at 2θ = 38.14°, 44.33°, 64.44° and 77.43° due to the Bragg reflections corresponding to the [111], [200], [220] and [311] sets of lattice planes. This may be indexed based on the fcc structure of Ag present in the composite. No spurious diffractions due to crystallographic impurities were found.49 The size of the Ag nanoparticles estimated from the Debye–Scherrer formula was 51.1 nm.50,51 The GO showed two peaks at 10.13° and 42.12° as depicted in Fig. 2Cb. The experimental diffraction angle of these peaks are found to be in good agreement with the values reported in literature.23,52
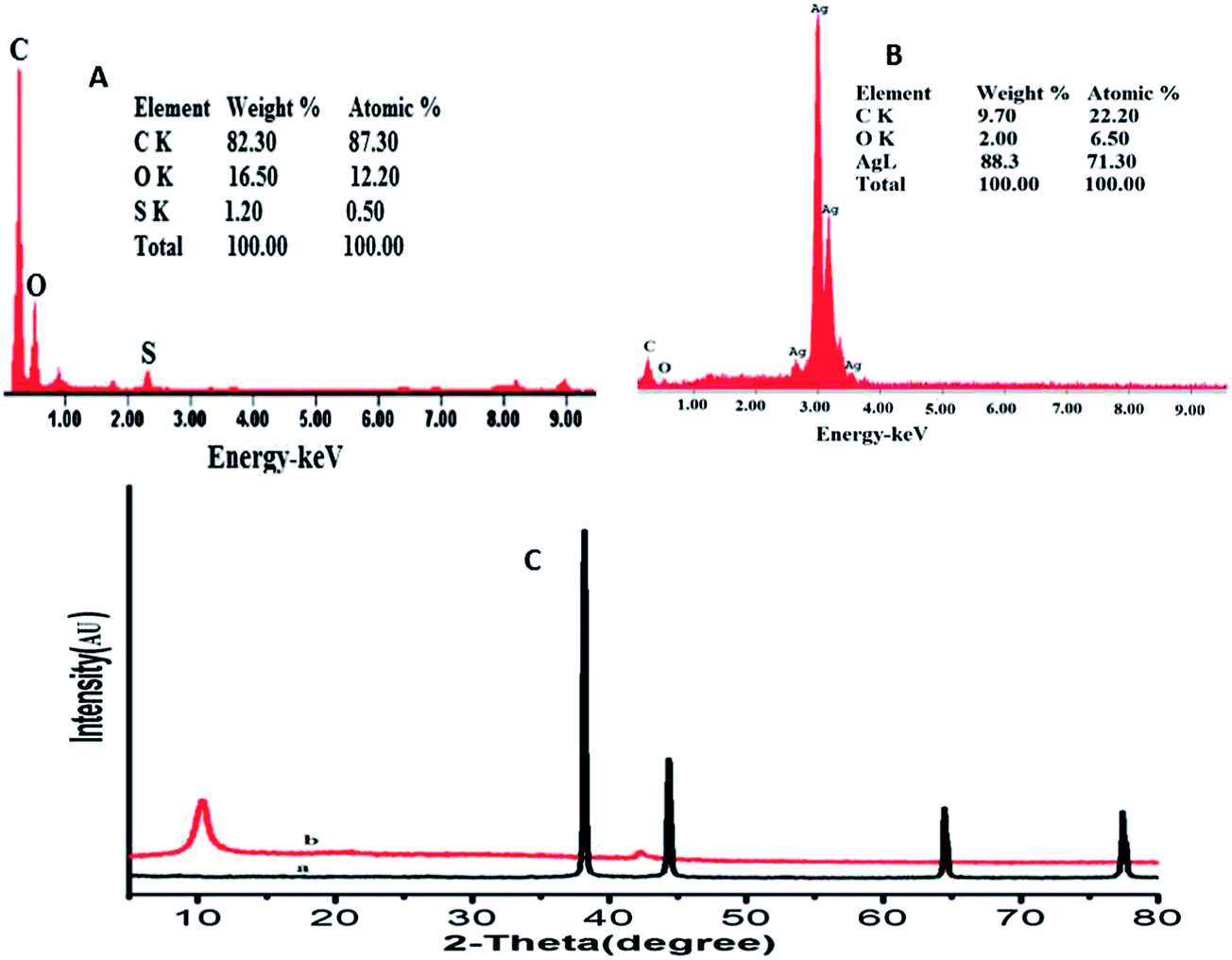 | ||
| Fig. 2 EDX spectra of GO film (A) and AgNPs–GO composite film (B). (C) XRD showing peak indices & 2θ positions of (a) AgNPs and (b) GO. | ||
Electrochemical characterization of AgNPs–GO/GCE
Electrochemical characterization of AgNPs–GO/GCE was carried out using electrochemical impedance spectroscopy (EIS) and chronocoulometry (CC). EIS is a useful method for probing the characteristics of a surface-modified electrode. The semicircle portion observed at high frequencies in the Nyquist diagrams corresponds to the charge transfer limiting process, and the charge transfer resistance (Rct) values can be directly measured as the diameter of the semicircle.53 The EIS results (Fig. S1, ESI†) show that the charge transfer resistance (Rct = 226 Ω) of AgNPs–GO/GCE is lower than that of GO/GCE (Rct = 259 Ω) and bare GCE (Rct = 276 Ω). This can be attributed to the excellent electrical properties and high surface area of GO and cyclodextrin stabilized silver nanoparticles, which acts as a fast electron conduction pathway between the electrode and the electrochemical probe.Electrochemically active surface areas were obtained by CC. The electrochemical effective surface area for bare GCE and modified electrodes was calculated by the slope of the plot of Q vs. t1/2 obtained by CC using 0.5 mM K3[Fe(CN)6] as a model complex (Fig. S2 ESI†) The electrochemical effective surface areas measured were 0.295 cm2 and 0.141 cm2, for AgNP–GO/GCE and GO/GCE, which are approximately 3.0 and 2.0 times the surface area of bare GCE (0.079 cm2), respectively.
Electrochemical behaviour of As(III) at nanostructured electrode
Cyclic voltammetry (CV) was used to investigate the electrocatalytic activity of AgNPs–GO/GCE for the electrochemical behaviour of As(III). Fig. 3 shows CVs at GCE, GO/GCE and AgNPs–GO/GCE in the absence of arsenic in 0.1 M H2SO4 at a scan rate of 100 mV s−1. There was no redox peak obtained at GCE and GO/GCE without arsenic, indicating the GO film was non electroactive in the selected potential region. The background current of GO/GCE was higher than that of GCE, which could be attributed to the large specific area of GO. However, a pair of redox peaks was observed at AgNPs–GO/GCE without arsenic (Fig. S3, ESI†). This can be attributed to the redox of Ag+/0 in the composite, in which the electroactive AgNPs are oxidized to Ag+ at 0.38 V vs. Ag/AgCl on the forward anodic scan, with conversion of Ag+ back to Ag at 0.24 V vs. SCE on the reversed cathodic scan.54 This also indicates that AgNPs have been successfully immobilized onto the GO surface. When 2.65 × 10−7 M As(III) was added into the 0.1 M H2SO4 solution, a weak pair of reduction and oxidation peaks at −0.12 and 0.16 V respectively vs. Ag/AgCl, was observed on the GO modified GCE, while no peak was observed at the bare GCE (Fig. 3B) indicating that GO can catalyze the reaction. However, when the AgNPs–GO composite was immobilized on the GCE surface, the reduction peak was significantly enhanced and the corresponding oxidation peak potential shifted negatively to 0.143 V vs. Ag/AgCl, and the associated oxidation peak current increased by a factor of three compared to GO/GCE (Fig. 3C). Hence, the synergistic effect of AgNPs and GO catalyses the electrochemical reaction of arsenic.55 This can be attributed to the good catalytic ability and, large surface area of AgNP–GO/GCE which facilitates the accumulation of the arsenic at the surface of the electrode and accelerates the electron transfer.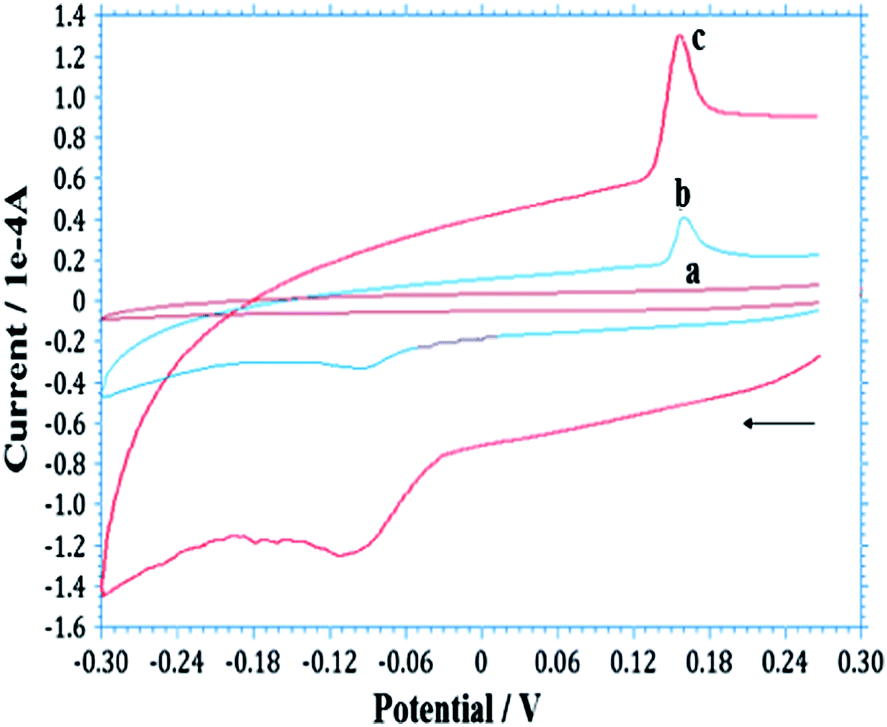 | ||
| Fig. 3 Typical CVs obtained for the As(III)/As(0) redox couple at (a) bare GCE, (b) GO/GCE and (c) AgNP–GO/GCE in 0.1 M H2SO4 solution containing 2.65 × 10−7 M As(III) at 100 mV s−1. | ||
The cyclic voltammetric study of As(III) at the AgNP–GO/GCE using different scan rates (v) in 0.1 M H2SO4 indicated that the reduction of As(III) to As (0) and the oxidation of As (0) to As(III) are surface-confined and diffusion-controlled processes, respectively. (Fig. S4 and S5 ESI†).
Anodic stripping voltammetry
Reproducible arsenic stripping peaks were only produced with H2SO4 as the supporting electrolyte. When HCl (1.0 M) was used as a supporting electrolyte, silver oxidation was observed (due to the possible formation of AgCl at the AgNPs surface) which strongly interfered with the As(III) signal.12,56 Various concentrations of H2SO4 (0.05, 0.1, 1.0 and 2.0 M) were explored for As(III) detection, and sensitivity decreased with respect to concentration. A concentration of 0.1 M was found as the best compromise between sensitivity and reproducibility. Thus, 0.1 M H2SO4 was selected as the supporting electrolyte for subsequent measurements. No obvious stripping peak was obtained for 3.99 × 10−7 M at bare GCE (Fig. S6a, ESI†). Under similar conditions, a stripping peak was observed when GO was on the electrode surface, and the peak was significantly enhanced when β-CD stabilized AgNPs were present on the surface (Fig. S6b and c, ESI†). The combined presence of β-CD stabilized AgNPs and GO thus provides a 3D network for the reduction of As(III) to As (0) during the deposition step. This shows that the reduction of As(III) to As (0) is more facile at the AgNPs–GO surface resulting in an increase in the stripping peak current. It is clear that As(III) was selectively deposited on the AgNPs–GO array. During pre-concentration, As(III) is adsorbed onto the AgNPs–GO modified electrode surface, then reduced (Scheme 1) and deposited on the electrode under cathodic potentiostatic conditions (−0.60 V) for 120 s. The high sensitivity of As(III) detection can be attributed to the presence of hydroxyl groups, which provide a large hydrophilic surface, and thus high adsorption capacity of β-CD stabilized AgNPs on the surface of GO.
 | ||
| Scheme 1 Schematic representation of β-CD-AgNPs–GO arsenic interaction and stripping out on modified GCE. Graphene oxide (GO), glassy carbon electrode (GCE), silver nanoparticles (AgNPs). | ||
Deposition of As(III) on the AgNPs–GO/GCE under optimized conditions (deposition potential: −0.6 V, frequency: 40 Hz, pulse amplitude: 25 mV, electrolyte: 0.1 M H2SO4) was carried out by using SW-ASV for various deposition times (1–5 min) (Fig. S7, ESI†). The area of the stripping peak (i.e. sensitivity) increased with the deposition time due to the increased amount of arsenic on the AgNPs–GO-modified GCE. The response of the modified electrode for As(III) (3.99 × 10−8 M) increased rapidly up to a 2 min deposition time and then remained relatively constant due to the limited remaining active sites. Increasing the deposition time improved the sensitivity and could be used to monitor low concentration levels.57 A 2 min deposition time was considered for subsequent experiments.
Detection performance of AgNPs–GO/GC electrode and limit of detection (LOD) under optimal conditions
The detection performance of the modified electrode was investigated using SW-ASV using the conditions discussed earlier. Fig. 4A shows typical SW-ASVs measured at the AgNPs–GO/GC electrode in 0.1 M H2SO4 solution containing various concentrations of As(III). The peak height of the stripping current signal increases linearly with the concentration of As(III) from 13.33 to 375.19 nM. From the linear plot of the peak height (ipa) vs. As(III) (plot 4B) and with the use of the regression equation ipa (μA) = 0.1805 (μM) + 0.997, the sensitivity and LOD of the sensor were estimated to be 180.5 μA μM−1and 0.249 nM (S/N = 3), respectively. These results were obtained by using the data points of five repeated measurements.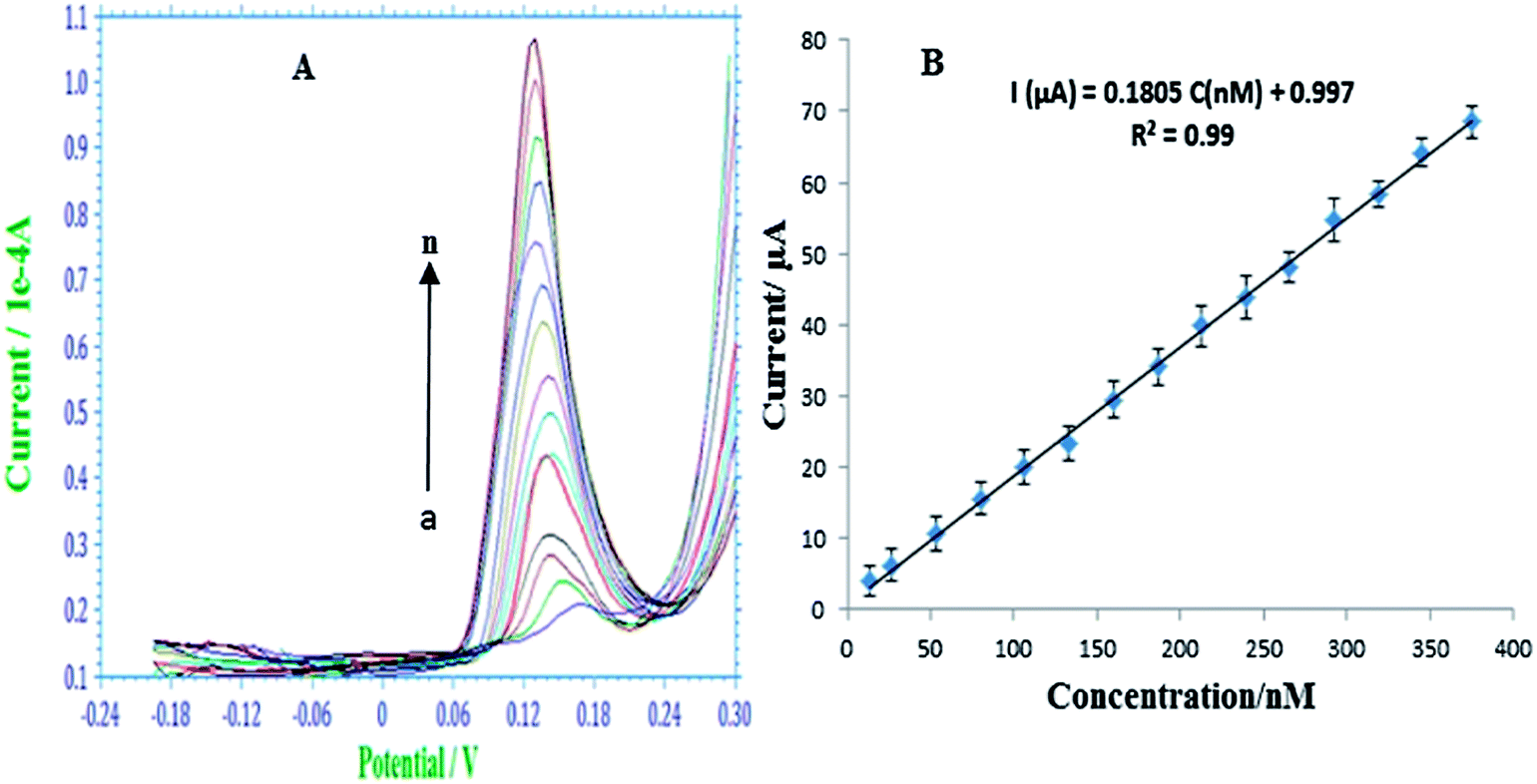 | ||
| Fig. 4 (A) SW-ASV responses with respect to increasing As(III) concentration, from a to n, 13.33, 26.65, 53.29, 79.72, 106.52, 133.11, 159.68, 186.23, 212.76, 239.28, 265.78, 292.26, 318.70, 345.10, 375.19 nM, respectively. Deposition potential: −0.60 V for 120 s, rest period: 10 s, frequency: 40 Hz, pulse amplitude: 25 mV. Data in the plot (B) represent the mean ± S.D. | ||
Various analytical parameters including electrolyte, measurement technique, detection range, sensitivity and LOD for the prepared AgNPs–GO/GC electrode are compared with recently reported values8,58–61 in Table 1. The detection limit, linear calibration range and sensitivity for arsenic determination at this modified electrode are comparable and even better than those obtained by using other modified electrodes based on metal or metal oxide nanoparticles.8,58–61
| Modified electrode | Electrolyte | Technique | Linear working rangea (nM) | Sensitivitya (μA μM−1) | LODa (nM) | Ref. |
|---|---|---|---|---|---|---|
| a Values were either tabulated directly from, or calculated by conversion used in, the corresponding references. | ||||||
| PtNPs/GCE | 1 M aqueous HClO4 | SW-ASV | 1000–50000 |
0.22 | 26.7 | 8 |
| Ir/BDDE | 1 mM phosphate buffer | CV | 93–9800 | 7.03 | 20.0 | 57 |
| AgNPs/CT/GCE | 0.1 M HNO3 | DPASV | 130–13300 |
0.308 | 16.0 | 58 |
| Au nano-array/GCE | 1 M H2SO4 | ASV | 9800–59200 |
0.91 | 80.0 | 59 |
| Nano-Pt-Fe(III)/MWCNT/GCE | 0.1 M H2SO4 | ASV | 100–10000 |
4.76 | 10.0 | 60 |
| AuNP–GCE | 1 M HCl | LSV | — | 14.2 | 1.0 | 61 |
| β-CD/AgNP–GO | 0.1 M H2SO4 | SW-ASV | 13.33–375.19 | 180.5 | 0.24 | Present work |
Simultaneous detection of As(III) and Cu(II)
Determination of As(III) in natural water samples is a challenging task because of co-deposition of other metals, particularly Cu(II), which is the main interferent during stripping analysis, reducing the sensitivity of the target ions.58 Determination of As(III) with varying concentrations of Cu(II) at the AgNPs–GO/GC electrode using SW-ASV is shown in Fig. 5. Two well-defined peaks are observed for As(III) and Cu(II) at 0.16 V and 0.05 V, respectively. It is observed from the figure that there is no interference of Cu(II) in this concentration range up to 3.75 × 10−7 M, thus, allowing out the method to simultaneously detect and quantify As(III) and Cu(II).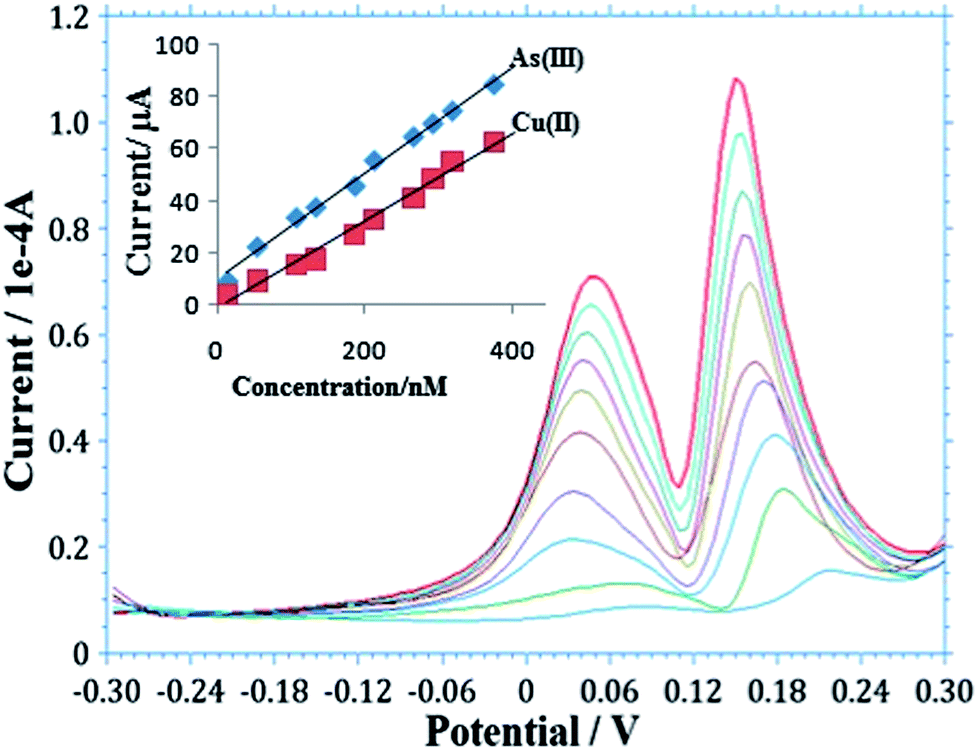 | ||
| Fig. 5 Simultaneous detection of As(III) and Cu(II) at AgNPs–GO/GCE using SW-ASV in 0.1 M H2SO4. (Insert: calibration plot for detection of As(III) and Cu(II). Other conditions are similar to Fig. 4). | ||
The calibration plot was obtained from the SW-ASV analysis for As(III) and Cu(II) (insert in Fig. 5). The corresponding linear fit for each metal represented by the equations,
| As(III): Y = 0.203X + 9.886 (n = 5, r2 = 0.933), |
| Cu(II): Y = 0.167X − 1.108 (n = 5, r2 = 0.985), |
Effect of other interferences
Interference of some inorganic ions and some organic materials on the stripping peak current of As(III) has been previously reported, especially in ground water samples.59 In the present study, we also tried to characterize the performance of the nanostructured AgNPs–GO/GC electrode in the presence of these materials. Table S1 (ESI†) reports the interference of some inorganic and organic materials on the stripping peak current of As(III) at different concentration levels. The effect on As(III) peak current caused by Na(I), Ca(II), Mg(II) ions was relatively small up to 3.33 × 10−3 M (NaCl, CaCl2, MgCl2) before an increase was observed, causing a corresponding potential shift and increase in the peak current. This may be due to the formation of AgCl on the electrode surface.12 Sulphate ions (Na2SO4, MgSO4 and CaSO4) did not interfere with the As(III) signal up to 1.33 × 10−4 M, while phosphate did not show significant interference up to 1.63 × 10−3 M.Interference on the As(III) signal due to ethylenediaminetetraacetate (EDTA), Triton X-100 and sodium dodecyl sulfate (SDS) was also tested. These materials can interfere with the As(III) signal due to complexation effects.59 However, in the present study, the presence of EDTA up to 6.67 × 10−3 M did not show any significant effect on As(III) detection. Also, no effect was caused by the addition of the Triton-X-100 (non-ionic surfactant) and the SDS (anionic surfactant) up to 1.33 × 10−3 M. Thus, the developed electrode showed good performance for As(III) detection in the presence of interfering substances, such as Cu(II) and some inorganic and organic molecules at the levels described herein.
Accuracy and precision
The accuracy expressed as a mean relative error of the developed method was determined by spiking with accurately weighed amounts (preanalyzed amounts) of arsenic. From our measurements, we determine a value of the mean percent recovery to be 99.63 and 100.11 in intraday and interday assays, respectively (Table S2, ESI†). These values give confidence in the accuracy of our method. The precision of the proposed procedure was estimated by analyzing arsenic in preanalyzed assay solutions for five times in intraday assay and four successive days for interday assay using the SW-ASV method. The percentage recovery and precision based on the average of five separate determinations are listed in ESI (Table S2†). The mean variation coefficient for intraday and interday assays was found to be 1.03% and 1.73%, respectively. The results confirm high precision of the proposed procedure and good stability of the electrode.Reproducibility and stability of the sensor
The reproducibility of the proposed approach was evaluated by intra- and inter-assay coefficients of variation. The intra-assay precision of this method was evaluated by assaying arsenic at two concentration levels for twenty replicative measurements using the same sensor, and the inter-assay precision was evaluated by assaying arsenic at two concentration levels for successive measurements using twenty independently made sensors. The intra-assay variation coefficients of this method were 4.2% and 3.7% at arsenic concentrations of 3.99 × 10−8 M and 7.98 × 10−8 M, respectively. The inter-assay variation coefficients at these concentrations were 3.9% and 4.5%, indicating that the sensor demonstrated good repeatability. The stability of the prepared sensor can be maintained by storing under a clean and dry condition. No obvious decrease in the response of arsenic was observed in the first 10 day storage. After a 90 day storage period, the sensor retained 94.1% of its initial current response. These results indicate that the sensor held satisfactory performances in reproducibility and stability, and can be confidently used for the analysis of arsenic in real samples.Determination of arsenic in real samples
The AgNPs–GO-modified GCE was used to detect arsenic in different water samples collected from ground water of Andheri (East), and Panchganga River, Mumbai, India (Fig. 6A and B). 10 ml of each sample was diluted to 100 ml with 0.1 M H2SO4 supporting electrolyte. 50 ml of sample was taken in a cell for detection of arsenic by the standard addition method. The measurements were performed five times. Panchganga River sample showed 4.84 × 10−8 M of As(III) while As(III) concentration in the ground water sample was below the detection limit of our method. Thus, a known quantity of 10.65 × 10−8 M of As(III) was added in ground water sample; the average value found was 10.42 × 10−8 M. Thus a recovery of 97.88% was obtained. This observation validates the suitability of As(III) detection in water samples using the developed sensor.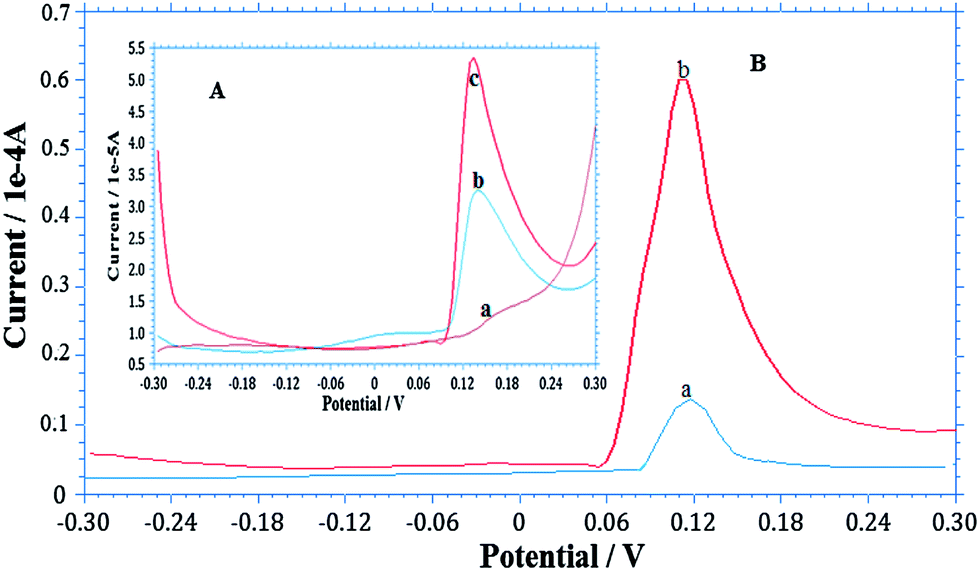 | ||
| Fig. 6 SW-ASV determination of As(III) in different water samples under optimized conditions similar to Fig. 4. (A) Ground water where insert Aa is voltammogram of ground water sample, Ab As(III) spiked and Ac is standard As(III) added. (B) River water, where Ba is voltammogram of river water sample and Bb is standard As(III) added. | ||
Conclusion
In summary, we used a green and facile approach to synthesize cyclodextrin stabilized AgNPs–GO composite and used it for the fabrication of a novel electrochemical sensor for As(III) detection using ASV. The developed sensor showed a low detection limit, good working range and its analytical characteristics outperformed most reported electrochemical sensors. We successfully used the AgNPs–GO composite sensor to detect As(III) in the presence of Cu and other organics. The results indicate that As(III) and Cu(II) could be determined simultaneously. The developed sensor was used to determine As(III) in ground and river water samples. The analysis showed a good recovery demonstrating the suitability of the material for the determination of As(III) in natural water and other As(III) containing samples.The synthesized AgNPs–GO composite has a number of attractive features, such as ease of synthesis, low toxicity, surface functionalities and excellent stability, which hold great promises for applications in other electroanalytical methods and for electro-catalysis.
Acknowledgements
The funding for this work is partly by the University Grants Commission, New Delhi, India under its Dr D. S. Kothari Post doctorate fellowship scheme (RAD) and partly by the US Army International Technology Center, Tokyo, Japan through contract number FA2386-12-1-4086. DPC acknowledges financial support from U.S. Army Research Laboratory (contract NNL-09AA00A). For TEM analysis, we acknowledge the support of the Maryland NanoCenter and its NispLab.References
- H. Teddy, F. Katia, E. David, C. Vincent, B. Philippe and G. Pierre, J. Electroanal. Chem., 2012, 664, 46–52 CrossRef PubMed.
- S. Abdollah, M. Hussein, H. Rahman and S. Saied, Sens. Actuators, B, 2008, 129, 246–254 CrossRef PubMed.
- F. Katia, Y. Vaiata, C. Bruno, G. Véronique and T. M. Danièle, Electrochem. Commun., 2010, 12, 1439–1141 CrossRef PubMed.
- H. Chien-Te, L. Chi-Yuan, C. Yu-Fu, L. Jiun-Sheng and T. Hsisheng, Carbon, 2013, 62, 109–116 CrossRef PubMed.
- J. Wang, Stripping Analysis: Principles, Instrumentation and Applications, CVH Publishers, Deerfield Beach, FL, 1985 Ch. 4 Search PubMed.
- D. Xuan, N. Olga, E. Michael Hyde and R. G. Compton, Anal. Chem., 2004, 76, 5924–5929 CrossRef PubMed.
- A. Salimi, M. E. Hyde, C. E. Banks and R. G. Compton, Analyst, 2004, 12, 99–114 Search PubMed.
- X. Dai and R. G. Compton, Analyst, 2006, 131, 516–521 RSC.
- A. I. Tribidasari, S. Rika, M. Yoshihiro, F. Akira and E. Yasuaki, Anal. Chem., 2006, 78, 6291–6298 CrossRef PubMed.
- D. Li, J. Li, X. Jia, Y. Han and E. Wan, Anal. Chim. Acta, 2012, 733, 23–27 CrossRef CAS PubMed.
- A. S. Julio, L. R. Bernabé, S. P. Amalia, B. Luis, P. Eduardo and I. Pignot-Paintrandb, Electrochim. Acta, 2010, 55, 4876–4882 CrossRef PubMed.
- A. O. Simm, C. E. Banks and R. G. Compton, Electroanalysis, 2005, 17, 1727–1733 CrossRef CAS PubMed.
- X. Ren, X. Meng, D. Chen, F. Tang and J. Jiao, Bioelectronics, 2005, 9, 433–437 CrossRef PubMed.
- A. Tiwari and S. K. Shukla, Advanced Carbon Materials and Technology, WILEY-Scrivener, USA, 2014 Search PubMed.
- A. Tiwari and M. Syvajarvi, Advanced Materials for Agriculture, Food and Environmental Safety, WILEY-Scrivener, USA, 2014 Search PubMed.
- J. Tyagi and R. Kakkar, Adv. Mater. Lett., 2013, 4, 721–736 Search PubMed.
- O. Parlak, A. Tiwari, A. P. F. Turner and A. Tiwari, Biosens. Bioelectron., 2013, 49, 53–62 CrossRef CAS PubMed.
- M. N. Muralidharan and S. Ansari, Adv. Mater. Lett., 2013, 4, 927–932 Search PubMed.
- H. Chen, M. B. Muller, K. J. Gilmore, G. G. Wallace and D. Li, Adv. Mater., 2008, 20, 3557–3561 CrossRef CAS PubMed.
- Q. Jian-Ding, H. Jing and L. Ru-Ping, Sens. Actuators, B, 2011, 160, 287–294 CrossRef PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney and E. A. Stach, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- C. C. Owen and T. N. Son Binh, Small, 2010, 6, 711–723 CrossRef PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett and G. Evmenenko, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- D. Li, M. B. Müller, S. Gilge, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- J. Zhou, M. Chen and G. W. Diao, ACS Appl. Mater. Interfaces, 2003, 5, 828 CrossRef PubMed.
- J. Zhou, M. Chen and G. W. Diao, J. Mater. Chem. A, 2013, 1, 2278 CAS.
- F. Liu, J. Y. Choi and T. S. Seo, Chem. Commun., 2010, 46, 2844 RSC.
- Y. J. Guo, S. J. Guo, J. T. Ren, Y. M. Zhai, S. J. Dong and E. K. Wang, ACS Nano, 2010, 4, 4001 CrossRef CAS PubMed.
- Y. J. Guo, S. J. Guo, J. Li, E. K. Wang and S. J. Dong, Talanta, 2011, 84, 60 CrossRef CAS PubMed.
- G. Yunlong, W. Bin, Y. M. Liu, Y. Ying, J. Zheng and G. Y. Liu, Adv. Mater., 2011, 40, 4626–4630 Search PubMed.
- B. Li, T. Liu, Y. Wang and Z. Wang, J. Colloid Interface Sci., 2012, 1, 114–121 Search PubMed.
- J. Li, D. Kuang, Y. Feng, F. Zhang, Z. Xu and M. Liu, J. Hazard. Mater., 2012, 201, 250–259 CrossRef PubMed.
- S. Park, K. S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–588 CrossRef CAS PubMed.
- N. Zhou, J. Li, H. Chen, C. Liao and L. Chen, Analyst, 2013, 138, 1091–1097 RSC.
- W. L. Zhang and H. J. Choi, Analyst, 2013, 138, 1091–1097 RSC.
- I. V. Lightcap, M. Sean, S. Timothy and P. V. Kamat, J. Phys. Chem. Lett., 2012, 3(11), 1453–1458 CrossRef CAS.
- A. Mukherjee, M. K. Sengupta and M. A. Hossain, J. Health Popul. Nutr., 2006, 24, 142–163 Search PubMed.
- R. C. William and J. R. Kenneth, Arsenic speciation in the environment, Chem. Rev., 1989, 89(4), 713–764 CrossRef.
- N. G. Debendra and U. B. Mazumder, J. Med. Sci., 2011, 9, 360–370 Search PubMed.
- Y. Mohammad, S. Nazmul, K. Samar and R. Mahfuzar, J. Med. Sci., 2011, 27, 371–376 Search PubMed.
- L. Gang, S. Guo-Xin, N. W. Paul, N. Luis and Z. Yong-Guan, Environ. Int., 2011, 37, 1219–1225 CrossRef PubMed.
- U. S. Environmental Protection Agency, National primary drinking water regulations contaminants monitoring, Fed. Regist., 2001, 66(14) Search PubMed.
- E. Munoz and S. Palmero, Talanta, 2005, 65, 613–620 CrossRef CAS PubMed.
- C. B. Hymer and J. A. Caruso, J. Chromatogr. A, 2004, 1045, 1–13 CrossRef PubMed.
- H. M. Van, C. Zhang, X. Zhang and R. Cornelis, Analyst, 2002, 127, 634–640 RSC.
- N. S. Gadhari, B. J. Sanghavi and A. K. Srivastava, Anal. Chim. Acta, 2011, 703, 31–40 CrossRef CAS PubMed.
- N. S. Gadhari, B. J. Sanghavi, S. P. Karna and A. K. Srivastava, Electrochim. Acta, 2010, 56, 627–635 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- R. P. Gopalan, Int. J. Nanosci., 2010, 9, 487–494 CrossRef.
- B. D. Cullity, Elements of X-ray Diffraction, Addison-Wesley Company, USA, 1956 Search PubMed.
- J. Rita and S. S. Florence, Chalcogenide Lett., 2009, 6, 269–273 Search PubMed.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176 CrossRef CAS PubMed.
- B. Y. Chang and S. M. Park, Anal. Chem., 2010, 3, 207–229 CAS.
- J. Li, D. Kuang, Y. Feng, F. Zhang, Z. Xu, M. Liu and D. Wang, Biosens. Bioelectron., 2013, 42, 198–206 CrossRef CAS PubMed.
- M. Welch, R. Christine and G. Compton, Anal. Bioanal. Chem., 2006, 3, 601 CrossRef PubMed.
- R. Feeney and S. P. Kounaves, Anal. Chem., 2000, 72, 2222 CrossRef CAS.
- T. A. Ivandini, R. Sato, Y. Makide, A. Fujishima and Y. Einaga, Anal. Chem., 2006, 78, 6291 CrossRef CAS PubMed.
- S. Prakash, T. Chakrabarty, A. K. Singh and V. K. Shahi, Electrochim. Acta, 2012, 72, 157–164 CrossRef CAS PubMed.
- R. Baron, B. Sljukic, C. Salter, A. Crossley and R. G. Compton, Russ. J. Phys. Chem. A, 2007, 9, 1443–1147 CrossRef.
- S. Seung-Hyun and H. Hun-Gi, Bull. Korean Chem. Soc., 2010, 11, 3077 Search PubMed.
- X. Dai, O. Nekrassova, M. E. Hyde and R. G. Compton, Anal. Chem., 2004, 76, 5924–5929 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra00934g |
| This journal is © The Royal Society of Chemistry 2014 |