
New insights into dihydrogenphosphate recognition with dirhenium(i) tricarbonyl complexes bridged by a thiourea moiety†
Abstract
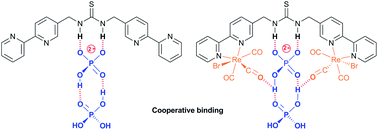
Three thiourea bridged 2,2′-bipyridine ligands bearing either a single thiourea group (L1), or two units separated by either a para (L2) or meta-substituted (L3) aromatic spacer, along with the corresponding bis(fac-tricarbonylrhenium(I)) complexes are reported. The three ligands all show the anticipated binding to acetate. However 1H NMR titrations reveal an unusual cooperative binding to, and selectivity for, two dihydrogenphosphate ions. The rhenium(I) complexes similarly demonstrate unusual sigmoidal titration curves, and in the case of {Re(CO)3Br}2(μ-L1) a surprisingly strong interaction to two anions. These were further exemplified in the emissive behaviour leading to the conclusion that there is an unusual interaction with dihydrogenphosphate, giving an initial increase in the emission, followed by a decrease and a blue shift in wavelength possibly as a result of partial deprotonation. It appears that dihydrogenphosphate binds cooperatively, with the addition of a second anion enhancing the interaction of the first, probably by proton transfer; this could explain the remarkable selectivity for phosphate seen with many reported anion receptors.
- This article is part of the themed collection: Supramolecular chemistry: self-assembly and molecular recognition
 Please wait while we load your content...
Please wait while we load your content...

