Binding mode prediction and identification of new lead compounds from natural products as renin and angiotensin converting enzyme inhibitors†
N. S. Hari Narayana Moorthy‡
*,
Natércia F. Brás‡,
Maria J. Ramos and
Pedro A. Fernandes*
REQUIMTE, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, 687, Rua do Campo Alegre, 4169-007, Porto, Portugal. E-mail: hari.moorthy@fc.up.pt; hari.nmoorthy@gmail.com; pafernan@fc.up.pt; Fax: +351-220 402 506; Tel: +351-220 402 506
First published on 15th April 2014
Abstract
Virtual screening studies were performed with docking and pharmacophore methods on a natural products (NPs) data set to investigate its inhibitory effect on cardiovascular targets such as renin (REN) and angiotensin-converting enzymes (ACE). Conformers obtained from the ligand–protein complex (from pdb) and the flexialigned structure of the reference compounds were used to generate pharmacophore query models. The results derived from the analysis on natural product data set showed that the compounds such as Nat-4, Nat-59, Nat-99 and Nat-141 provided good results against REN and Nat-7, Nat-6, Nat-31, Nat-59 and Nat-61 are considered as good HITs against ACE. The present studies revealed that the compound Nat-59 has remarkable interaction with both the studied targets (ACE and REN). Also we have observed better effects for those compounds on the hERG target through the pharmacophore based virtual screening method, revealing that these can be used as antiarrythmic agents. A new lead compound (NLC-1) was designed on the basis of the pharmacophoric features of the selected HITs, provided good HIT properties by pharmacophore and docking studies. Molecular dynamics simulations were performed on the compound NLC-1 in order to find out the binding features of this molecule on the targets (ACE and REN). These results concluded that the natural compound identified from these studies and the designed compound (NLC-1) can have multiple activities on cardiovascular targets.
Introduction
Cardiovascular diseases are one of the leading causes of death in the world. Hypertension is one of the leading risk factors in cardiovascular diseases (congestive heart failure, stroke, myocardial infarction, etc.) and this chronic condition affects nearly 25% of adults worldwide. The current therapies available for this disease mainly act on the Renin–Angiotensin–Aldosterone System (RAAS) which plays a primordial role in blood pressure regulation and electrolyte homeostasis (being involved in the pathogenesis of hypertension and several renal diseases). However, the antihypertensive drugs available do not cure the disease completely, further approximately 70% of those people with hypertension do not reach their target blood pressure levels.1–4The RAAS pathway has primary role in controlling blood pressure through the production of angiotensin-II (AT-II) from angiotensin-I (AT-I) by the action of angiotensin-I converting enzyme (ACE).5 Initially, the RAAS proteolytic cascade starts with the release of the enzyme renin (REN) (EC 3.4.23.15) by the juxtaglomerular cells located in the kidney. REN activates a plasma protein named angiotensinogen (452 residues) by hydrolyzing the peptidic bond between 10th and 11th positions, releasing a decapeptide known as AT-I. Finally, the inactive AT-I is further cleaved into the octapeptide AT-II by the metalloprotease ACE.2,6 ACE hydrolyzes both the inactive AT-I into vasoconstrictor AT-II and the vasodilator bradykinin into an inactive metabolite leading to blood pressure up-regulation.5,7,8
The REN and ACE play important role for the production of the vasoconstrictor AT-II and the inhibitors discovered for these targets are used for the treatment of hypertension. However, the inhibition of ACE causes REN overexpression, which in part compensates the effect of inhibiting ACE and AT-I functions.4 The inhibition of REN is much more attractive, it catalyzes the rate-limiting step of this enzymatic cascade and angiotensinogen is the only known substrate. Therefore, drugs that inhibit REN are expected to have several advantages and they should be an attractive antihypertensive strategy with fewer side effects.5,9–11 The peptidomimetic REN inhibitor, for example, remikiren (RO 42-5892) was discovered during the 1980s. More recent research has lead to the discovery of several classes of non-peptidic REN inhibitors.12,13 In 2007, the Food and Drug Administration (FDA) approved the first and unique direct REN inhibitor, aliskiren (commercialized by Novartis as Tekturna). The first breakthrough in the area of ACE inhibitor was captopril, which was launched in 1981 for hypertension management.14–16
The development of novel class of antihypertensive drugs include the development of single compound capable of inhibiting more than one enzymatic activity simultaneously involved in hypertension pathophysiology is important nowadays.5,17 Hence, in the present investigation, we have used some computational methods to discover novel antihypertensive agents from natural molecules (natural products (NPs)). NPs from different structural classes have been investigated for its action on REN and ACE enzymes. NPs traditionally have played an important role in drug discovery and were the basis of most early medicines.18–20
However, in the drug discovery research, still there is a shortage of lead compounds progressing into clinical trials. This is especially in the therapeutic areas such as oncology, immunosuppression and metabolic diseases where NPs have played a central role in lead discovery. In the world market, NPs and NP-derived drugs were well represented in the top 35 selling ethical drugs in between 2000 and 2002.18,21 Hence, the virtual screening analysis of NPs for the therapeutic targets provides significant lead for the design of novel molecules. In the present investigation, we have performed virtual screening of NPs on REN and ACE targets using computational techniques (pharmacophore, docking, molecular dynamic (MD) simulations and correlation analysis) (Table S1†). On the basis of the study results, we have designed a novel lead compound for further development.
Experimental
Virtual screening (docking study)
The X-ray crystallographic structures of REN and ACE (2V0Z and 1O86 codes at 2.2 Å and 2.0 Å of resolution, respectively) were obtained from the Protein Data Bank.22,23 These REN and ACE enzymes are complexed with their well-known inhibitors aliskiren and lisinopril respectively. All crystallographic water molecules and inhibitors were removed to obtain the unbound structures of each enzyme. All hydrogen atoms were added, taking into account of all residues in their physiological protonation state. Structures of all studied ligands were built with GaussView 5.0 software. Ligand–protein docking was performed with the vsLab (virtual screening lab),24 an easy-to-use graphical interface for the molecular docking software AutoGrid/AutoDock that has been included into VMD as a plug-in. AutoDock 4 (ref. 25) was used for the docking calculations, and the program VMD 1.9.1 (ref. 26) was used for visual inspection, analysis and preparation of the figures of the docking results. Kollman partial charges were assigned to all protein atoms, while for the zinc (Zn2+) and the chloride ions (Cl−), formal charges of +2 and −1 respectively were applied. The grid maps were centred into inhibitors and comprised 52 × 52 × 51 points of 0.375 Å spacing in REN, as well as 55 × 55 × 55 points of 0.375 Å spacing in ACE. The Lamarckian genetic algorithm27 was employed with the following parameters: population size of individuals: 150; maximum number of energy evaluations: 2.5 × 106 and maximum number of generations: 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. For all the calculations, 50 docking rounds were performed with the step sizes of 2.0 Å for translations and with orientations and torsions of 5.0°. The ΔGbind values of the NPs were obtained from the scoring function of AutoDock program that is based on the Amber force field. Docked conformations were clustered within 2.0 Å root-mean-square-deviations (rmsd) to prevent similar poses, and a further visual analysis was also used to eliminate unfavourable poses and interactions.
000. For all the calculations, 50 docking rounds were performed with the step sizes of 2.0 Å for translations and with orientations and torsions of 5.0°. The ΔGbind values of the NPs were obtained from the scoring function of AutoDock program that is based on the Amber force field. Docked conformations were clustered within 2.0 Å root-mean-square-deviations (rmsd) to prevent similar poses, and a further visual analysis was also used to eliminate unfavourable poses and interactions.
Virtual screening (pharmacophore based)
The ligand based (flexialigned reference compounds) and the complex based (protein–ligand complex from pdb) pharmacophore analyses were used to investigate the structural features responsible for the interactions and further virtual screening of NP compounds. The pharmacophore analysis of the data set (above mentioned) was carried out using MOE software.28 For ligand based pharmacophore analysis, initially, the conformers of the reference compounds were developed by stochastic search covering maximum number of conformers generated to 1000, superpose RMSD to 0.15 Å and the fragment strain limit of 4 kcal mol−1. The lowest energy conformers obtained from the stochastic search was aligned using ligand flexibility on MMFF94x force field with the energy cutoff of 10 Å for non-bonded interactions. The following properties have been calculated for the aligned structures: the strain energy (U), the mutual similarity score (F) and the value of the objective function (S) of each alignment. The aligned structures exhibit lower U, F and S values were considered for the pharmacophore analysis.The details of the pharmacophore queries used for the pharmacophore based virtual screening studies are given below. The pharmacophore contours and its radius used in the pharmacophore query models development are provided in Table 1.
| Pharmacophores | Code | Contour | Radius |
|---|---|---|---|
| Pharmacophore-1 | F1 | Acc | 2 |
| F2 | Acc | 1.5 | |
| F3 | ML|Acc2 | 1.5 | |
| F4 | Aro|Hyd | 2 | |
| F5 | Don | 1 | |
| Pharmacophore-2 | F1 | Acc | 1 |
| F2 | Acc | 1 | |
| F3 | ML|Acc2 | 1.5 | |
| F4 | Acc | 1.3 | |
| F5 | Aro|Hyd | 1.4 | |
| Pharmacophore-3 | F1 | Ani|Acc|Acc2|Don2 | 2.5 |
| F2 | Cat|Acc2|Don | 1.5 | |
| F3 | Acc|Don2 | 2.5 | |
| F4 | Acc2|Don | 1.5 | |
| F5 | Hyd|Acc2|Don2 | 2.5 | |
| +V1 h | Excluded volume | 1.5 | |
| Pharmacophore-4 | F1 | Ani|Acc|Acc2|Don2 | 2 |
| F2 | Hyd|Acc | 1 | |
| F3 | Hyd | 1.5 | |
| F4 | Aro|Hyd|Acc|Don | 1.5 | |
| F5 | Hyd|Acc2|Don2 | 1.5 | |
| +V1 h | Exterior volume | 1.35 | |
| Pharmacophore-5 | F1 | Don | 1.5 |
| F2 | Don | 1.5 | |
| F3 | Hyd|Aro | 1.3 | |
| F4 | Hyd | 1.5 |
(1) In complex based pharmacophore analysis, the protein–ligand complexes obtained from protein data bank were used. The PDB structures such as 1O86 and 2V0Z were utilized for ACE (pharmacophore-1) and REN (pharmacophore-3) respectively.
(2) The flexialigned structure of the reference compounds such as captopril, enalapril, lisinopril and rimipril for ACE (pharmacophore-2) and compounds such as aliskiren, enalkiren, rimikiren and zankiren for REN (pharmacophore-4) were used to create pharmacophore query models.
(3) In order to analyze the effect of the molecules on hERG target, a pharmacophore query was generated with the inhibitor present in pdb 3O0U (pharmacophore-5).29
Molecular dynamics simulations protocol
All hydrogen atoms were added to both REN:NLC-1 and ACE:NLC-1 complexes with the Amber software X-Leap, taking into account of all residues in their physiological protonation state. The only exception was the proton donor residue (Asp38) of REN enzyme, which was protonated. Seven and fourteen counter-ions (Na+) were employed to neutralize the high negative charges of REN and ACE systems respectively. The X-Leap program was also used for this purpose. An explicit solvation model with pre-equilibrated TIP3P water molecules was used, filling a truncated octahedral box with a minimum 12 Å distance between the box faces and any atom of the protein. The size of these two systems was 16944 and 23597 atoms. To calculate the optimized geometries and electronic properties for the subsequent parameterization of NLC-1, the Gaussian09 suite of programs30 was used to perform restricted Hartree–Fock calculations, with the 6-31G(d) basis set. The methodology chosen was consistent with that adopted for the parameterization process in the AMBER 10.0 software.31 The atomic charges were calculated using the RESP algorithm.32 All geometry optimizations and MD simulations were performed with the parameterization adopted in AMBER 10.0 simulations package, using the Amber 2003 force field (parm03)33,34 for the proteins and the GAFF force field35 for the compound NLC-1. The initial geometry optimization of both systems occurred in two stages, in order to release the bad contacts in the starting structures. Initially, the protein was kept fixed and only the position of the water molecules and counter-ions was minimized (500 steps using the steepest descent algorithm and 1500 steps carried out using conjugate gradient). In the second stage, the full system was minimized (5000 steps using the steepest descent algorithm and 10000 steps were carried out using conjugate gradient). Subsequently, an MD simulation of 50 ps at constant volume and temperature, and considering periodic boundaries conditions was run, followed by 6 ns of MD simulation with an isothermal–isobaric ensemble for each system in which Langevin dynamics was used (collision frequency of 1.0 ps−1) to control the temperature at 310.15 K.36 All simulations presented here were carried out using the PMEMD module, implemented in the Amber 10.0 simulations package.31 Bond lengths involving a hydrogen bond were constrained using the SHAKE algorithm, and the equations of motion were integrated with a 2 fs time-step using the Verlet leapfrog algorithm.37 The Particle-Mesh Ewald (PME) method38 was used to include the long-range interactions and the non-bonded interactions were truncated with a 10 Å cutoff. The MD trajectory was saved every 2 ps and the MD results were analyzed with the PTRAJ module of AMBER 10.0.31
Correlation studies
A data set comprised of structurally varying REN and ACE inhibitors possessing inhibitory constant values (Ki in μM) were collected from the binding database (http://www.bindingdb.org). After removing the duplicate compounds in the dataset using MOE software, 86 and 63 compounds as REN and ACE inhibitors respectively, were converted into the ΔGbind by using the formula ΔGbind = RTlnKi. In order to carry out the correlation analysis, the data set was spilt into training and test set compounds (almost 25–30% of the compounds were treated as test set) by random selection using Statistica software (Statistica (8.0), StatSoft Inc. Tulsa, OK, USA, 2010). For each of the targets, two test sets were used for the analysis and the ΔGbind of the test set was calculated from the developed models. Leave one out (LOO) crossvalidation analysis was performed to investigate the predictive ability of the developed models. Furthermore, the ΔGbind values for the natural compounds were calculated using the developed correlation models.
Results and discussion
The NPs data set was undergone virtual screening by docking and pharmacophore based methods. The results derived from the studies are discussed here.Virtual screening studies (docking)
In order to validate the docking method that was used in this study, initially we have performed docking of the well-known inhibitor aliskiren to the REN active site, as well as the lisinopril, captopril and enalaprilat inhibitors to the ACE active site. The root-mean-square deviation (RMSD) values of the best predicted docking solutions in relation to their crystallographic positions and the ΔGbind values of each inhibitor associated to its respective enzymes are used to evaluate our results. Concerning REN enzyme, the RMSD value obtained for the docking of aliskiren is 1.38 Å, showing a ΔGbind value of −11.49 kcal mol−1, which vary 1.09 kcal mol−1 from the experimental value (−12.58 kcal mol−1).39 These two small variations reveal the higher ability of our protocol to predict the binding mode of ligands into the REN active site. Regarding the ACE enzyme, the RMSD values obtained for lisinopril, enalaprilat and captopril are 2.23 Å, 0.90 Å and 0.57 Å, respectively, which indicate a higher proximity to their X-ray geometries. The presence of more conformations for each inhibitor probably will reduce this RMSD value. However, this will be very time and resource consuming and is not expected to provide any relevant improvement. In addition, small variations are observed between the experimental values and our computational ΔGbind values obtained for these three complexes: lisinopril (−11.75 kcal mol−1 vs. −12.48 kcal mol−1), enalaprilat (−11.18 kcal mol−1 vs. −12.97 kcal mol−1) and captopril (−10.71 kcal mol−1 vs. −9.89 kcal mol−1).40 Hence, all these small variations also reveal a higher ability of our protocol to predict the binding mode of ligands in ACE active site. Fig. 1 shows the superposition of inhibitors in the X-ray structure of REN and ACE proteins and our docking solutions. It is observed a very similar arrangement of all inhibitors inside the enzyme active sites. | ||
| Fig. 1 The superimposition of the docked inhibitors (coloured by element and depicted ball and stick representation) and the respective X-ray structures (coloured green and depicted ball and stick representation). | ||
Subsequently, the docking parameters used in the initial studies executed correctly the binding mode and affinity of these known compounds. Hence, we have applied the same docking protocol to evaluate the binding interactions of all NPs data set compounds studied. The binding energy and inhibitory constant values obtained for the first twenty ligands are shown in Table 2.
| REN | ACE | ||||||
|---|---|---|---|---|---|---|---|
| Inhibitor | Ki (μM) | ΔGbind (kcal mol−1) | Inhibitor | Ki (μM) | ΔGbind (kcal mol−1) | ||
| Docking | Correlation analysis | Docking | Correlation analysis | ||||
| Nat-20 | 1.46 × 10−2 | −10.69 | −9.1704 | Nat-34 | 7.10 × 10−4 | −12.48 | −10.3527 |
| Nat-38 | 1.94 × 10−2 | −10.52 | −9.1704 | Nat-7 | 7.72 × 10−4 | −12.43 | −10.3527 |
| Nat-39 | 4.15 × 10−2 | −10.07 | −9.1703 | Nat-6 | 1.20 × 10−3 | −12.17 | −10.3527 |
| Nat-59 | 3.06 × 10−2 | −10.25 | −9.1703 | Nat-31 | 6.82 × 10−3 | −11.14 | −10.3523 |
| NLC-1 | 6.33 × 10−2 | −9.82 | −9.1702 | Nat-59 | 7.54 × 10−3 | −11.08 | −10.3523 |
| Nat-165 | 6.43 × 10−2 | −9.81 | −9.1702 | NLC-1 | 1.38 × 10−2 | −10.72 | −10.352 |
| Nat-7 | 7.36 × 10−2 | −9.73 | −9.1702 | Nat-61 | 2.11 × 10−2 | −10.47 | −10.3516 |
| Nat-19 | 8.43 × 10−2 | −9.65 | −9.1701 | Nat-69 | 2.26 × 10−2 | −10.43 | −10.3515 |
| Nat-132 | 8.72 × 10−2 | −9.63 | −9.1701 | Nat-72 | 2.34 × 10−2 | −10.41 | −10.3515 |
| Nat-14 | 1.63 × 10−1 | −9.26 | −9.1697 | Nat-10 | 2.46 × 10−2 | −10.38 | −10.3515 |
| Nat-12 | 1.77 × 10−1 | −9.21 | −9.1697 | Nat-47 | 2.81 × 10−2 | −10.3 | −10.3512 |
| Nat-77 | 2.21 × 10−1 | −9.08 | −9.1695 | Nat-165 | 2.86 × 10−2 | −10.29 | −10.3512 |
| Nat-15 | 2.28 × 10−1 | −9.06 | −9.1694 | Nat-38 | 2.96 × 10−2 | −10.27 | −10.3512 |
| Nat-99 | 2.28 × 10−1 | −9.06 | −9.1694 | Nat-39 | 3.45 × 10−2 | −10.18 | −10.3509 |
| Nat-141 | 2.32 × 10−1 | −9.05 | −9.1694 | Nat-40 | 3.50 × 10−2 | −10.17 | −10.3509 |
| Nat-23 | 2.53 × 10−1 | −9 | −9.1693 | Nat-67 | 3.69 × 10−2 | −10.14 | −10.3508 |
| Nat-24 | 2.66 × 10−1 | −8.97 | −9.1693 | Nat-19 | 4.08 × 10−2 | −10.08 | −10.3506 |
| Nat-81 | 2.89 × 10−1 | −8.92 | −9.1692 | Nat-24 | 4.08 × 10−2 | −10.08 | −10.3506 |
| Nat-22 | 2.89 × 10−1 | −8.92 | −9.1692 | Nat-68 | 5.25 × 10−2 | −9.93 | −10.35 |
| Nat-163 | 2.99 × 10−1 | −8.9 | −9.1691 | Nat-99 | 5.25 × 10−2 | −9.93 | −10.35 |
| Nat-82 | 3.25 × 10−1 | −8.85 | −9.1690 | Nat-15 | 5.43 × 10−2 | −9.91 | −10.3499 |
REN
The presence of a hairpin (generally called flap) is a common feature to all aspartic proteases. Therefore, the flap of REN is composed by the residues Thr72 to Ser81 and it covers the central part of the REN binding site. Fig. 2 shows 3D representations of REN, enhancing the catalytic dyad (Asp38 and Asp226), the flap residues coloured at green and the main non-polar residues around the active site (coloured at dark blue) that are responsible for substrate/inhibitor binding. | ||
| Fig. 2 Representation of REN enzyme, with both catalytic Asp residues in ball and sticks, the flap residues in green and the main non-polar residues around the active pocket in dark blue. | ||
The database used in the present study has 165 natural compounds, which are described in ESI in Table S1.† We have considered only the five top-ranked inhibitor molecules for further discussion to simplify our results. The natural compounds Nat-20, Nat-38, Nat-39, Nat-59 and Nat-165 are the best five inhibitors bound to REN active site (structures provided in Table 3). Their representations and the most important interactions established within the active site of REN are shown in Fig. 3. It has observed that the major interactions within the pocket site of REN occurred by hydrophobic and van der Waals (vdW) interactions, as well as strong hydrogen bridges established with both catalytic aspartate residues. The residues that interact with the natural compounds are the following: Val36, Asp38, Tyr83, Thr85, Pro118, Phe119, Phe124, Val127, Asp226, Ala229, Tyr231 and Met303 (residue number provided as per the human REN sequence number). The compound Nat-20 establishes a very short H-bridge (1.88 Å) with the negative charged Asp226 by its hydroxyl group, whilst the compound Nat-38 establishes two important H-bonds with the carboxylic group of catalytic Asp38 (2.37 Å) and the carbonyl side chain group of Thr85 (2.28 Å). Other strong hydrogen bridges are also established between a carbonyl and hydroxyl groups of compound Nat-39 to the carboxyl group of Asp38 (1.84 Å) and the OH group of Tyr231. Compounds Nat-59 and NLC-1 possess several hydroxyl groups, which establish several relevant H-bonds with the carboxylic groups of Asp38 and Asp226, the carbonyl side chain of Thr85 and the carbonyl backbone of Pro118. The oxygen atom in Nat-165 interacts with the carboxylic side chain of Asp38 by an H-bonding (3.17 Å). This oxygen atom is less reactive than the hydroxyl and carbonyl groups present in other molecules, which is probably the reason for its weakest binding energy when compared with the previously described compounds. In addition, the aromatic side chains of residues Tyr83, Phe119, Phe124 and Tyr231 make several vdW interactions and T-shaped π–π stacking interactions with different aromatic moieties present in these five NPs. Similar hydrophobic contacts also occurred between the Val36, Val127, Pro118 and Met303 residues to some non-polar groups of all five-ranked inhibitors.
| Comp. name | Target | Structures |
|---|---|---|
| Nat-4 | ACE |  |
| Nat-6 | REN, ACE |  |
| Nat-7 | REN, ACE |  |
| Nat-10 | REN |  |
| Nat-20 | REN |  |
| Nat-31 | ACE |  |
| Nat-34 | ACE |  |
| Nat-38 | REN |  |
| Nat-39 | REN |  |
| Nat-59 | REN, ACE |  |
| Nat-61 | ACE |  |
| Nat-71 | REN |  |
| Nat-99 | REN, ACE |  |
| Nat-141 | REN |  |
| Nat-165 | REN |  |
| NLC-1 | ACE, REN |  |
 | ||
| Fig. 3 Representation of the interactions established by the residues of REN enzyme to the best five-ranked inhibitors: compounds Nat-20, Nat-38, Nat-39, Nat-59, Nat-165 and NLC-1. | ||
In fact, the binding free energy values and binding pose of these five-ranked inhibitors are very similar (a difference of 0.88 kcal mol−1 between the ΔGbind of the first and fifth compound), which indicates the crucial importance of REN residues such as Val36, Asp38, Tyr83, Thr85, Pro118, Phe119, Phe124, Val127, Asp226, Ala229, Tyr231 and Met303 interact with these natural compounds.
The binding interaction derived for the NPs identical with the reported compounds in the literatures. In the reported literatures, Asp38 and Asp226 residues in REN make hydrogen bonding interactions with the hydrogen bond acceptor or donor groups present in the molecules. Depending upon the nature of the inhibitors present in the active site, the residues such as Tyr14, Arg74, Tyr75, Ser76, Tyr77, Phe112, Gln128, Ala218, etc. are predominantly interacted with the molecules by hydrogen bonds or π–CH interactions or CH–CH or π–π stacking interactions.41–44
ACE
As previously mentioned, ACE (EC 3.4.15.1) is a zinc metallopeptidase that belongs to the M2 gluzincin family. There are two isoforms such as somatic (sACE) and testicular (tACE) available for this enzyme. The tACE is found in germinal cells and is a single monomer composed of 701 residues (Fig. 4). Since the ACE is a zinc dependent enzyme, it is expected that the best inhibitors bound the active site by strong interactions with this cation (Zn2+). According to the ΔGbind values obtained by docking study, the best five inhibitors are Nat-34, Nat-7, Nat-6, Nat-31 and Nat-59. The Ki values of the known ACE inhibitors lisinopril, enalaprilat and captopril are 2.4 nM, 6.3 nM and 14 nM, respectively,40 which indicate that the lisinopril is the most potent ACE inhibitor. Comparing its Ki value with the predicted values obtained for the top ranked NPs, it was verified that Nat-34 (0.71 nM), Nat-7 (0.77 nM) and Nat-6 (1.2 nM) show predicted potency 3, 3 and 2 times higher than lisinopril. Furthermore, the predicted potency of the NLC-1 compound (13.8 nM) is similar to the captopril drug (14 nM). Extended analyses are required to confirm these compounds as effective antihypertensive agents. All these facts highlight the importance of this data and suggest that these natural compounds could be a starting point for a new generation of antihypertensive drugs.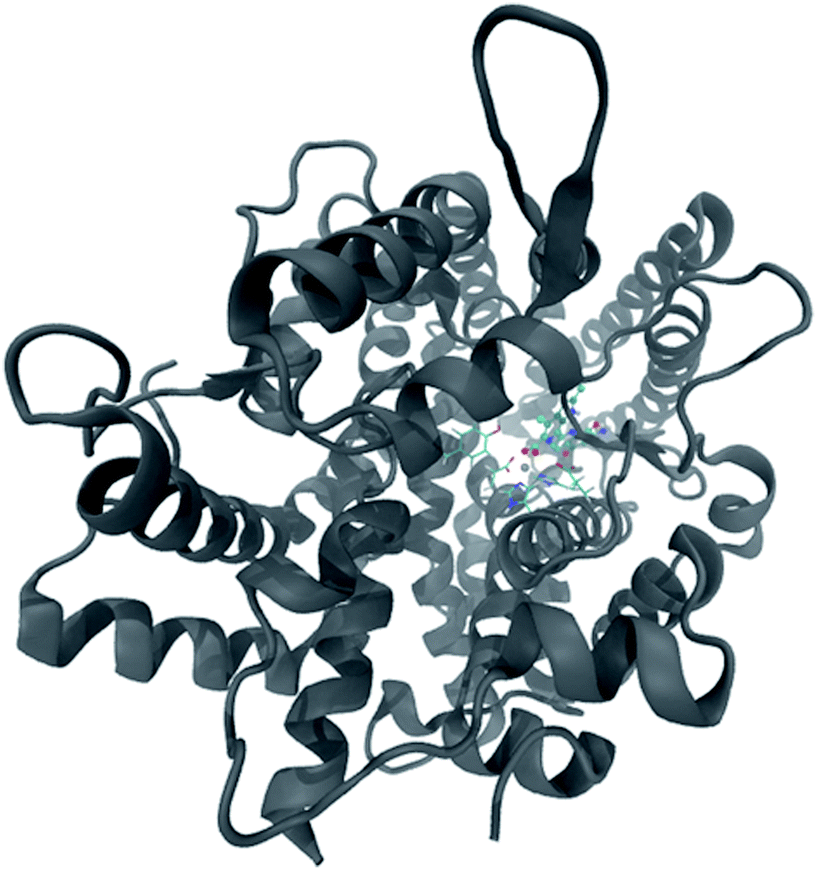 | ||
| Fig. 4 Representation of ACE enzyme with its catalytic site residues in sticks. | ||
Fig. 5 shows the interactions established between these compounds and the residues present in the binding pocket of this enzyme. The compound Nat-34 is the one that establish more interactions with ACE protein. As seen, the NH2 group makes a hydrogen bond with the hydroxyl group of Tyr360, a carbonyl group establish hydrogen bonds with the Tyr394, Glu403, His410 and a NH group interacts with the backbone carbonyl of Ala356. Its carboxylate group strongly interacts with the Zn2+ ion as well as with the OH group of Tyr523. In addition, the benzene ring possesses π–π stacking contacts with the aromatic side chain of Phe391 and Phe512 residues. The second-ranked inhibitor establishes H-bridges with CO and NH backbone groups of Ser355 and Ala356, respectively, as well as hydrophobic contacts with the Phe391. This compound also has a carboxylate group that coordinates in a bidentated form to Zn2+ ion. Compound Nat-6 interacts with Asp358, Ala356 and Tyr523 by H-bonds, as well as its COO− group is bidentated to Zn2+. The binding mode of compound Nat-31 is very similar to the compound Nat-34. Thus, it interacts with Tyr360, Tyr394, His410, Glu403 and Tyr523 by hydrophilic contacts, while the aromatic residues Phe391, Phe512, Tyr523 and His410 make hydrophobic contacts. Similarly to the REN enzyme, the compounds Nat-59 and NLC-1 also interact well with this metalloprotease (ACE). It makes T-shaped π–π stacking interactions with Phe512 and Tyr523; hydrophilic contacts with Asn70, Ser355, Lys368, Glu384 and Tyr520; and bidentated coordination with Zn2+ ion by one hydroxyl group and an oxygen-ring atom. All these five compounds show similar interactions with the ACE active site, which agrees with the proximity of their binding mode conformations and binding energies values (a difference of 1.76 kcal mol−1 between the binding energies of the first and fifth compounds). Therefore, the main hydrophilic amino acids those contribute to an efficient inhibitor binding are the following: Asn70, Ser355, Asp358, Lys368, His383, Glu384, His387, Glu403, His410, Glu411 and Glu485; whilst the crucial aromatic residues are the Tyr360, Phe391, Tyr394, Phe512, Tyr520 and Tyr523.
 | ||
| Fig. 5 Representation of the interactions established by the residues of ACE enzyme to the best five-ranked inhibitors: compounds Nat-34, Nat-7, Nat-6, Nat-31, Nat-59 and NLC-1. | ||
The binding interaction analysis of different ACE inhibitors on the catalytic site of N and C-domain of the ACE reported on the literature showed that the residues such as Gln281, His353, His513, Tyr520 and Tyr523 are predominantly interacted with the inhibitors in C-domain ACE. However, in the N-domain ACE, the residues such as Gln259, His331, His491, Tyr498 and Tyr501 are present in the active site for the interaction. This shows that mainly the same amino acid residues present in both the domain of the enzyme for the interaction.45 The study performed by Tzakos showed that the residues such as Gln281, His353, His513, Tyr520 and Tyr523 importantly present for the ligand interaction. They reported that the interactions governing ACE_C domain and the ligand are: a salt bridge between Asp377 and Glu162, and the NH2 group, a hydrogen bond of the inhibitor with Gln281, and a π–π stacking interaction of Phe391 with aromatic group. In ACE_N, inhibitors interact with Glu431, Tyr369 and Arg381 by hydrogen bonds and a salt bridge between the carboxy group in the inhibitors and Arg500. Some specific residues also present in different compounds for the interaction.45–47 The results derived from the present study also coincided with the reported results in the literature. In our studies His383, His387, Glu384, Phe391, Tyr394, His410, Glu411, Phe512, Tyr520 and Tyr523 are preferably interacted with the inhibitors.
Pharmacophore based virtual screening
In this pharmacophore analysis, two queries each for REN and ACE have been generated using flexialigned reference compounds and the conformers present in the protein–ligand complexes derived from the protein data bank (2V0Z and 1O86 for REN and ACE respectively). The pharmacophore contour features and their volume (radius) utilized in these analysis are provided in Table 1. Importantly, the hydrophobic (hyd), hydrogen bond acceptor (Acc), projected hydrogen bond acceptor (Acc2), aromatic/hydrophobic (Aro/Hyd), anionic atom (Ani), cationic atom (Cat), projected hydrogen bond donors (Don2), metal ligator (ML) and hydrogen bond donor (Don) features were considered as query pharmacophore contours (Fig. 6). The excluded and exterior volumes of 1.5 Å and 1.35 Å were used for the receptor and reference REN pharmacophores. | ||
| Fig. 6 Pharmacophore models used for the virtual screening. | ||
The pharmacophore-1 (receptor ACE) and the pharmacophore-2 (reference ACE) developed with 5 pharmacophore contours features. The pharmacophore-3 (receptor REN) and the pharmacophore-4 (reference REN) also possessed 5 contour features and a volume parameter such as excluded volume in pharmacophore-3 and exterior volume in pharmacophore-4 models. The generated pharmacophore queries (pharmacophores 1–4) have been used to perform virtual screening, in order to derive HIT molecules from the NPs data set. The detail of the significant HIT compounds obtained from the pharmacophore based virtual screening study is given in Tables 4 and 5. Among the 165 NP compounds present in the data set, 6 compounds are identified as significant compounds through pharmacophore-1 and 16 compounds identified through pharmacophore-2. It is interesting that the HITs identified through the pharmacophore-1 are also present in the pharmacophore-2. The compounds Nat-61, Nat-59, Nat-33, Nat-82, Nat-3 and Nat-31 are considered as significant HITs from pharmacophore pharmacophore-1 and 2. These compounds exhibited the RMSD values ≤1. The HITs identified through pharmacophore-3 and 4 (REN) are provide in Table 5. Those pharmacophore query models provided four compounds each as significant HITs. It is interesting that the compound Nat-59 is selected as one of the significant compounds against the REN target. This compound possessed the RMSD value <2 for both the queries (pharmacophore-3 and 4). This compound (Nat-59) has been selected as significant HIT through all the pharmacophore queries (1–4) for REN and ACE.
| Comp. code | RMSD | |
|---|---|---|
| Pharmacophore-1 | Pharmacophore-2 | |
| Nat-61 | 0.8804 | 0.8541 |
| Nat-59 | 0.8867 | 0.8470 |
| Nat-33 | 0.9612 | 0.7533 |
| Nat-82 | 0.9814 | 0.7771 |
| Nat-3 | 1.0569 | 0.7465 |
| Nat-31 | 1.0586 | 0.6173 |
| Nat-4 | — | 0.4865 |
| Nat-6 | — | 0.5959 |
| Nat-34 | — | 0.6091 |
| Nat-60 | — | 0.7440 |
| Nat-27 | — | 0.7932 |
| Nat-133 | — | 0.8294 |
| Nat-24 | — | 0.9210 |
| Nat-45 | — | 0.9335 |
| Nat-7 | — | 0.9600 |
| Nat-44 | — | 0.9788 |
| NLC-1 | 0.8743 | 0.6832 |
| Comp. code | RMSD | |
|---|---|---|
| Pharmacophore-3 | Pharmacophore-4 | |
| Nat-10 | 1.6178 | — |
| Nat-4 | 1.6857 | — |
| Nat-59 | 1.7001 | 1.2119 |
| Nat-99 | 1.4345 | — |
| Nat-141 | — | 1.0065 |
| Nat-46 | — | 1.1549 |
| Nat-71 | — | 1.0943 |
| NLC-1 | 1.7600 | 1.3456 |
The virtual screening results (docking and pharmacophore studies) on the REN and ACE showed that some compounds possessed significant docking score and those compounds also identified as good HITs through the pharmacophore analysis. Compounds such as Nat-4, Nat-59, Nat-99 and Nat-141 are considered as good compounds through both methods against REN target. The compounds Nat-7, Nat-6, Nat-31, Nat-59 and Nat-61 are considered as good HITs against ACE. These derived virtual screening results on REN and ACE by docking and pharmacophore analyses describe that the compound Nat-59 is a significant compound against both targets. Structure of the significant compounds selected from the studies is provided in Table 3.
The compound especially Nat-59 has significant effect on the active site of REN and ACE. This has been confirmed by the docking and pharmacophore analyses. Also the compounds such as Nat-6, Nat-7, Nat-38, Nat-39 and Nat-99 also possessed interaction with REN and ACE targets. The pharmacophore analysis also showed that Nat-31 has been identified as good HIT for ACE. On the basis of the structural description and pharmacophore features of Nat-59, Nat-31, Nat-6, Nat-38 and Nat-34, we have designed a new lead compound against ACE and REN targets. According to docking results, the developed new compound NLC-1 has significant binding free energy (−10.25 kcal mol−1) comparing to the reference compound aliskiren (−11.49 kcal mol−1) on REN target. In ACE target, NLC-1 possessed the binding free energy of −11.08 kcal mol−1, which is comparable to the reference ACE inhibitors. The pharmacophore analysis (pharmacophore 1–4) also selected this compound as significant HIT, the RMSD value of the compound is provided in Tables 4 and 5. This confirms that NLC-1 has significant interaction with REN and ACE targets. The interaction of NLC-1 revealed that mode of interactions is same as Nat-59 against both targets (Fig. 2 and 4).
We have also performed pharmacophore analysis of the NPs against one of the cardiovascular target human ether-a-go-go related gene (hERG). The pharmacophore analysis performed on the compounds showed that 72 compounds were identified as HITs for the target. It is interesting that those compounds identified as significant compounds from docking and pharmacophore analysis for ACE and REN inhibitory effects also considered as significant hit. The structural features of these molecules are comparatively similar to the features needed for the hERG blocking activities. Earlier reports on hERG blockers showed that the flexibility of the molecules orients the substituent towards the active site. Low polarizable groups and aromatic rings (hydrophobicity) provide optimum vdW surface properties required for the hERG blocking activity. The identified HITs also possessed flexible bonds, aromatic rings and polarizable groups in their structure.48–50 Hence, these compounds have significant interactions with the hERG protein and can used as antiarrythmic agents. The compound NLC-1 has been considered as significant hit for all cardiovascular targets. The conformers selected through the pharmacophore based virtual screening analysis are provided in Table 6.
| Pharmacophore | Conformers |
|---|---|
| Pharmacophore 1 |  |
| Pharmacophore 2 |  |
| Pharmacophore 3 |  |
| Pharmacophore 4 |  |
| Pharmacophore 5 |  |
MD simulations analysis
Furthermore, the molecule NLC-1 has undergone molecular dynamic simulation on REN and ACE enzymes. We have studied the conformational rearrangements of REN and ACE in complex with NLC-1 by MD simulations of 6 ns on each complex. Fig. 7 shows the RMSD values for protein backbone and for the compound NLC-1. As shown, the backbone values range between 1.5 Å and 2.0 Å during the MD simulations, indicating the stability of the folding and secondary structure of both proteins. Although the RMSD values obtained for NLC-1 compound is small in both complexes and is more tightly bound to the REN protein during the simulation due to its smallest value of ca. 0.5 Å. | ||
| Fig. 7 RMSD values obtained for the backbone of REN (black) and ACE (red) proteins, as well as for the compound NLC-1 bound to REN (green) and ACE (blue). | ||
Fig. 8 shows the RMSF values obtained for the REN and ACE bound enzymes by residue. Concerning to REN, the residues such as Leu1-Thr6, Gln150-Lys154, Glu167-Ser171, Glu187-Phe190, Val210-Thr214, Gly262-Pro266 and Tyr290-Lys294 show the highest RMSF values and possess high mobility, which probably may promote an easier inhibitor binding. It is expected that all these key residues will assist the flap-opening and closure to allow access for the substrate to the REN active pocket, enabling its catalytic reaction. The compound NLC-1 occupies the active site and the flap is stably closed over part of it, the RMSF values for the flap (residues 75–85) are indeed low (approximately 0.6 Å). This fact agrees with the higher stabilization provided by the establishment of several and strong interactions between the NLC-1 and the enzymatic active site.
 | ||
| Fig. 8 RMSF values obtained for REN:NLC-1 (blue) and ACE:NLC-1 (red) complexes. | ||
Similarly analyzing the RMSF values for ACE, it was observed higher values for residues Val1-Lys9, Asn68-Asn72, His116-Ser120, Lys137-Trp143, Ala212-Pro234, Val254-Pro260, Ser398-Asn408, Thr440-Asn443, Cys459-Asp468, Gln502-Pro509, Ser518-Glu520 and Lys560-Pro586. Since, these groups are the ones that mostly modify their position during the simulation studied, that probably help for the binding of substrate/inhibitor to the enzymes.
In order to examine the parts (atom or groups) of compound (NLC-1) have significant flexibility, the RMSF values were also calculated for this molecule when it is bound to REN and ACE. Fig. 9 shows these RMSF values and the numeration of all heavy atoms of NLC-1. It is possible to observe several peaks that have RMSF values above 0.5 Å. The regions of NLC-1 with the highest RMSF values correspond to several hydroxyl groups, which have higher flexibility during the MD simulation, due to their rotatable bonds that have high conformational flexibility to establish H-bonds with the active pocket residues of REN and ACE.
 | ||
| Fig. 9 RMSF values obtained for NLC-1 compound by atom bound to REN (blue) and ACE (red), as well as the representation of its 2D structure with the respective numeration of the heavy atoms. | ||
Structural information on the REN and ACE binding sites, in particular the H-bonds and vdW contacts established between the NLC-1 and the neighbour residues during the MD simulations were also analyzed. It is noteworthy that the NLC-1 compound has mainly hydrophobic character due to its aromatic rings, but it also possesses several hydroxyl groups. Fig. 10 shows the structures that are closest to the average structures of the REN:NCL-1 and ACE:NLC-1 complexes. In relation to REN, it is verified that several hydrophobic residues (Val36, Pro118, Ala122, Val127, Ala229, Leu224, Ile305 and Ala314) establish dispersive contacts with the NLC-1 and may be responsible for its efficient binding. It is also observed a T-shaped π–π stacking contacts between the NLC-1 rings and the aromatic side chains of Tyr83, Phe119 and Phe124 residues, as well as H-bonds between some hydroxyl groups of this molecule and the backbone NH groups of Ser84 and Gly86 (5% and 9% of occupancies), the backbone NH and OH groups of Thr85 (46% and 61% of occupancies), the OH groups of Ser230 and Thr309 (28% and 11% of occupancies) and the unprotonated carboxylic group of Asp226 with an average distance of 1.83 ± 0.45 Å and 62% of occupancy.
 | ||
| Fig. 10 Representation of the structures that is closest to the average structures of the REN:NLC-1 (left) and ACE:NLC-1 (right) complexes. | ||
The binding pocket of ACE is a narrow tunnel highly populated by aromatic residues such as Trp318, Tyr484, His371, Phe418, Phe473 and His474, which establish strong π–π stacking contacts with the NLC-1 aromatic rings. These non-polar groups of NLC-1 also made dispersive contacts with the non-polar Val341 and Val479 residues of ACE. However, the most important contact of this compound with the ACE active site has its direct and strong interaction with the metallic Zn2+ cation with an average distance of 2.00 ± 0.38 Å. Furthermore, several H-bonds are established between the OH groups of NLC-1 compound and the amino groups of Gln242 (14% of occupancy), Arg483 (28% of occupancy), His371 (10% of occupancy) and Lys472 (5% of occupancy), the negatively charged carboxylic group of Glu337 (89% of occupancy), the OH groups of Tyr480 and Tyr484 (8% and 7% of occupancies) and the backbone carbonyl group of Ala315 (86% of occupancy).
Our results highlight the importance, for the inhibitor efficiency, with the short and strong H-bonds, dispersive and π–π stacking contacts, demonstrating that all these interactions contribute to an efficient binding of NLC-1 to the REN and ACE active sites, and allow the stabilization of the active site scaffold of these two complexes.
Radial distribution functions in REN:NLC-1 and ACE:NLC-1 complexes
To evaluate the presence of water molecules and the solvation spheres in which they pack around the active site residues (Asp38 and Asp226 of REN; His344, Glu345, His348 and Glu372 of ACE), the radial distribution function (RDF) and the number of water molecules accumulated in their first sphere for the two systems were also determined (Fig. 11 and 12). From the Fig. 11 and 12, it is possible to observe that all catalytic residues only have one water molecule around. This occurs due to the presence of the compound NLC-1, which prevents the entrance of other conserved catalytic water molecules (involved in the nucleophilic attack during both catalytic reactions) into the active site. | ||
| Fig. 11 RDF (full line) and number of water molecules (dashed line) around the Asp38 and Asp226 residues. These values were measured between the oxygen atom of water molecules and the oxygen atoms of the carboxylic groups of Asp38 and Asp226 residues. | ||
 | ||
| Fig. 12 RDF (full line) and number of water molecules (dashed line) around the His344, Glu345, His348 and Glu372 residues. These values were measured between the oxygen atom of water molecules and the oxygen atoms of the carboxylic (Glu345 and Glu372) and amine (His344, His348) groups. | ||
Correlation analysis
In order to investigate the correlation between the ΔGbind and the inhibitory constant (Ki), 86 and 63, REN and ACE inhibitors respectively, from the binding database were collected. Correlation analyses were performed on all the compounds (model 1 for REN and model A for ACE) and two training sets (with almost 70–75% compounds) (models 2 and 3 for REN and Model B and C for ACE). The statistical parameters calculated from the analysis are provided in Table 7 and it showed that the developed models have the correlation coefficient values (R) >0.6 and the Ftest and ttest values are significant at 99% and 99.9% confidence levels respectively. The correlation between the ΔGbind and the Ki are graphically represented in Fig. 13. The validation analysis performed with internal method (leave one out (LOO)) and external (test set) showed that the crossvalidated correlation coefficients (Q2) of the models are significant (>0.4). The predicted ΔGbind values of the molecules using the models are provided in Tables S2 and S3† and graphically represented in Fig. 14. Furthermore, the ΔGbind of the NPs were also calculated and the predicted ΔGbind values are provided in Tables 2 and S4.†| Statistical Parameters | REN | ACE | ||||
|---|---|---|---|---|---|---|
| Model 1 | Model 2 | Model 3 | Model A | Model B | Model C | |
| a R = correlation coefficient, Q2 = crossvalidated correlation coefficient, LOO = leave one out. | ||||||
| Number of compounds | 86 | 55 | 63 | 63 | 46 | 43 |
| R values | 0.6405 | 0.7027 | 0.6825 | 0.6003 | 0.7038 | 0.6710 |
| Ftest | 58.4207 | 51.6971 | 53.1902 | 34.3710 | 43.1951 | 33.5855 |
| Ttest | −35.38 | −32.24 | −30.16 | −38.5000 | −39.85 | −35.02 |
| Q2LOO | 0.5116 | 0.4981 | 0.5139 | 0.6309 | 0.4872 | 0.4658 |
| Q2test | — | 0.5024 | 0.4625 | — | 0.5455 | 0.4165 |
 | ||
| Fig. 13 Correlation plot between Ki (μM) and ΔGbind (kcal mol−1) of REN and ACE inhibitors. | ||
 | ||
| Fig. 14 Scatterplot of ΔGbind (kcal mol−1) (experimental vs. predicted) of REN and ACE inhibitors from the correlation studies. | ||
In summary, it has verified that the active sites of both REN and ACE enzymes possess predominantly hydrophilic charged amino acids (mainly negatively) such as Glu and Asp and aromatic residues (Phe, Tyr, etc.) with hydrophobic characteristics. Therefore, the main interactions between all inhibitors and both enzymatic classes occurred by hydrogen bonds, hydrophobic π–π stacking and very close contacts with the Zn2+ ion (for ACE). The pharmacophore queries developed for the pharmacophore based virtual screening also showed predominantly hydrophilic contours such as Acc, Acc2, Don, Don2, etc. and aromatic contour (Aro/Hyd). These query contours also coincided with the binding mode studies performed with the docking analysis. Herein, comprehensive molecular docking and pharmacophore studies about the binding mode of several NPs into the REN and ACE active sites were analysed.
The results obtained indicate that the scoring function of AutoDock is adequate in predicting the correct binding modes of well-known inhibitors in relation to their available crystallographic structures. Subsequently, similar protocol was applied to correctly describe the binding mode of several NPs into the REN and ACE active sites. Hence these analysis results such as pharmacophore and docking analysis provide active site characters of REN and ACE. Our results suggest that the best compound to inhibit both enzymatic targets is the compound Nat-59. The designed compound NLC-1 also has the same binding interaction with both targets as Nat-59. The MD simulations performed on the NLC-1 with both the enzymes also confirm that they bind with the same residues in the enzymes. The new compound shares the pharmacophore of Nat-59, Nat-6, Nat-7, Nat-31 and Nat-34. Therefore, these findings are particularly relevant for the design of novel compounds and these ligand and structure-based approaches may lead to improved inhibitors.
Acknowledgements
Two of the Authors (N. S. H. N. Moorthy and N. F. Brás) gratefully acknowledge the Fundação para a Ciência e Tecnologia (FCT-Portugal) for their Postdoctoral Grants (SFRH/BPD/44469/2008 and SFRH/BPD/71000/2010, respectively) and Project PTDC/QUI-QUI/121744/2010.References
- D. D. Holsworth, M. Jalaie, T. Belliotti, C. Cai, W. Collard, S. Ferreira, N. A. Powell, M. Stier, E. Zhang, P. McConnell, I. Mochalkin, M. J. Ryan, J. Bryant, T. Li, A. Kasani, R. Subedi, S. N. Maitib and J. J. Edmunds, Bioorg. Med. Chem. Lett., 2007, 17, 3575 CrossRef CAS PubMed.
- N. F. Bras, M. J. Ramos and P. A. Fernandes, Phys. Chem. Chem. Phys., 2012, 14, 12605 RSC.
- O. Bezençon, D. Bur, T. Weller, S. Richard-Bildstein, L. Remen, T. Sifferlen, O. Corminboeuf, C. Grisostomi, C. Boss, L. Prade, S. Delahaye, A. Treiber, P. Strickner, C. Binkert, P. Hess, B. Steiner and W. Fischli, J. Med. Chem., 2009, 52, 3689 CrossRef PubMed.
- L. Coates, H. F. Tuan, S. Tomanicek, A. Kovalevsky, M. Mustyakimov, P. Erskine and J. Cooper, The catalytic mechanism of an aspartic proteinase explored with neutron and X-ray diffraction, J. Am. Chem. Soc., 2008, 130(23), 7235 CrossRef CAS PubMed.
- R. Fernández-Musoles, J. B. Salom, D. Martínez-Maqueda, J. J. López-Díez, I. Recioe and P. Manzanares, Food Chem., 2013, 139, 994 CrossRef PubMed.
- X. Jeunemaitre, A. P. Gimenez-Roqueplo and J. Celerier, Curr. Hypertens. Rep., 1999, 1, 31 CrossRef CAS PubMed.
- R. M. Carey and H. M. Siragy, Endocr. Rev., 2003, 24, 261 CrossRef CAS PubMed.
- B. Williams, J. Hypertens., 2009, 27(suppl. 3), S19 CrossRef CAS PubMed.
- N. F. Brás, P. A. Fernandes and M. J. Ramos, J. Biomol. Struct. Dyn., 2014, 32, 351–363 Search PubMed.
- C. Jensen, P. Herold and H. R. Brunner, Nat. Rev. Drug Discovery, 2008, 7, 399 CrossRef CAS PubMed.
- A. Politi, S. Durdagi, P. Moutevelis-Minakakis, G. Kokotos, M. G. Papadopoulos and T. Mavromoustakos, Eur. J. Med. Chem., 2009, 44, 3703 CrossRef CAS PubMed.
- C. M. Tice, Z. Xu, J. Yuan, R. D. Simpson, S. T. Cacatian, P. T. Flaherty, W. Zhao, J. Guo, A. Ishchenko, S. B. Singh, Z. Wua, B. B. Scott, Y. Bukhtiyarov, J. Berbaum, J. Mason, R. Panemangalore, M. G. Cappiello, D. Müller, R. K. Harrison, G. M. McGeehan, L. W. Dillard, J. J. Baldwin and D. A. Claremon, Bioorg. Med. Chem. Lett., 2009, 19, 3541 CrossRef CAS PubMed.
- C. M. Tice, Annu. Rep. Med. Chem., 2006, 41, 155 CAS.
- S. Hanessian, S. Guesne, L. Riber, J. Marin, A. Benoist, P. Mennecier, A. Rupin, T. J. Verbeurenc and G. D. Nanteuilb, Bioorg. Med. Chem. Lett., 2008, 18, 1058 CrossRef CAS PubMed.
- M. A. H. Ismail, M. N. Aboul-Enein, K. A. M. Abouzid, D. A. A. El Ella and N. S. M. Ismail, Bioorg. Med. Chem., 2009, 17, 3739 CrossRef CAS PubMed.
- E. M. Gordon, J. D. Godfrey, H. N. Weller, S. Natarajan, J. Pluscec, M. B. Rom, K. Niemela, E. F. Sabo and D. W. Cushman, Bioorg. Chem., 1986, 14, 148 CrossRef CAS.
- B. Battistini, P. Daull and A. Y. Jeng, Cardiovasc. Drug Rev., 2005, 23, 317 CrossRef CAS.
- M. S. Butler, Nat. Prod. Rep., 2005, 22, 162 RSC.
- D. J. Newman, G. M. Cragg and K. M. Snader, Nat. Prod. Rep., 2000, 17, 215 RSC.
- A. D. Buss, B. Cox and R. D. Waigh, in Burger's Medicinal Chemistry and Drug Discovery, Sixth Edition, Volume 1: Drug Discovery, ed. D. J. Abraham, Wiley, Hoboken, New Jersey, 2003, p. 847 Search PubMed.
- M. Butler, J. Nat. Prod., 2004, 67, 2141 CrossRef CAS PubMed.
- J. Rahuel, V. Rasetti, J. Maibaum, H. Rüeger, R. Göschke, N. C. Cohen, S. Stutz, F. Cumin, W. Fuhrer, J. M. Wood and M. G. Grütter, Chem. Biol., 2000, 7(7), 493 CrossRef CAS.
- R. Natesh, S. L. U. Schwager, E. D. Sturrock and K. R. Acharya, Nature, 2003, 421(6922), 551 CrossRef CAS PubMed.
- N. Cerqueira, N. J. Ribeiro, P. A. Fernandes and M. J. Ramos, Int. J. Quantum Chem., 2011, 111(6), 1208 CrossRef CAS.
- N. Cerqueira, P. A. Fernandes and M. J. Ramos, Biophys. J., 2006, 90(6), 2109 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14(1), 33 CrossRef CAS.
- G. M. Morris, D. S. Goodsell, R. Huey and A. J. Olson, J. Comput.–Aided Mol. Des., 1996, 10(4), 293 CrossRef CAS.
- MOE, Chemical Computing Group Inc., Montreal, H3A 2R7, Canada, 2011.
- Z. Rankovic, J. Cai, J. Kerr, X. Fradera, J. Robinson, A. Mistry, W. Finlay, G. McGarry, F. Andrews, W. Caulfield, I. Cumming, M. Dempster, J. Waller, W. Arbuckle, M. Anderson, I. Martin, A. Mitchell, C. Long, M. Baugh, P. Westwood, E. Kinghorn, P. Jones, J. C. Uitdehaag, M. van Zeeland, D. Potin, L. Saniere, A. Fouquet, F. Chevallier, H. Deronzier, C. Dorleans and E. Nicolai, Bioorg. Med. Chem. Lett., 2010, 20, 6237 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, M. Crowley, C. W. Ross, W. Zhang, K. M. Merz, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin and P. A. Kollman, AMBER 10, University of California, San Francisco, 2008 Search PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97(40), 10269 CrossRef CAS.
- D. A. Case, T. E. Cheatham 3rd, T. Darden, H. Gohlke, R. Luo, K. M. Merz Jr, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26(16), 1668 CrossRef CAS PubMed.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang and P. Kollman, J. Comput. Chem., 2003, 24(16), 1999 CrossRef CAS PubMed.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25(9), 1157 CrossRef CAS PubMed.
- J. A. Izaguirre, D. P. Catarello, J. M. Wozniak and R. D. Skeel, J. Chem. Phys., 2001, 114(5), 2090 CrossRef CAS PubMed.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23(3), 327 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103(19), 8577 CrossRef CAS PubMed.
- H. Tzoupis, G. Leonis, G. Megariotis, C. T. Supuran, T. Mavromoustakos and M. G. Papadopoulos, J. Med. Chem., 2012, 55(12), 5784 CrossRef CAS PubMed.
- N. Dimitropoulos, A. Papakyriakou, G. A. Dalkas, E. D. Sturrock and G. A. Spyroulias, J. Chem. Inf. Model., 2010, 50(3), 388 CrossRef CAS PubMed.
- R. L. Webb, N. Schiering, R. Sedrani and J. Maibaum, J. Med. Chem., 2010, 53, 7490 CrossRef CAS PubMed.
- R. Goschke, S. Stutz, V. Rasetti, N. C. Cohen, J. Rahuel, P. Rigollier, H. P. Baum, P. Forgiarini, C. R. Schnell, T. Wagner, M. G. Gruetter, W. Fuhrer, W. Schilling, F. Cumin, J. M. Wood and J. Maibaum, J. Med. Chem., 2007, 50, 4818 CrossRef PubMed.
- Y. Wu, C. Shi, X. Sun, X. Wu and H. Sun, Bioorg. Med. Chem., 2011, 19, 4238 CrossRef CAS PubMed.
- Y. Nakamura, C. Sugita, M. Meguro, S. Miyazaki, K. Tamaki, M. Takahashi, Y. Nagai, T. Nagayama, M. Kato, H. Suemune and T. Nishi, Bioorg. Med. Chem. Lett., 2012, 22, 4561 CrossRef CAS PubMed.
- N. Dimitropoulos, A. Papakyriakou, G. A. Dalkas, E. D. Sturrock and G. A. Spyroulias, J. Chem. Inf. Model., 2010, 50, 388 CrossRef CAS PubMed.
- A. G. Tzakos and I. P. Gerothanassis, ChemBioChem, 2005, 6, 1089 CrossRef CAS PubMed.
- X. Wang, S. Wu, D. Xu, D. Xie and H. Guo, J. Chem. Inf. Model., 2011, 51, 1074 CAS.
- N. S. H. N. Moorthy, M. J. Ramos and P. A. Fernandes, Curr. Drug Targets, 2013, 14(1), 102 CrossRef CAS.
- N. S. H. N. Moorthy, M. J. Ramos and P. A. Fernandes, SAR QSAR Environ. Res., 2012, 23, 521 CrossRef CAS PubMed.
- N. S. H. N. Moorthy, M. J. Ramos and P. A. Fernandes, Curr. Drug Discovery Technol., 2012, 9, 25 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra00856a |
| ‡ Both authors are equally contributed in this work. |
| This journal is © The Royal Society of Chemistry 2014 |