Enhanced hydrogen storage properties of LiAlH4 catalyzed by CoFe2O4 nanoparticles†
Ziliang Lia,
Fuqiang Zhaib,
Qi Wana,
Zhaojiang Liua,
Jiawei Shana,
Ping Li*a,
Alex A. Volinskyc and
Xuanhui Qua
aInstitute for Advanced Materials and Technology, University of Science and Technology Beijing, Beijing 100083, China. E-mail: ustbliping@126.com; Fax: +86-10-62334311; Tel: +86-10-82377286
bDepartament Física Aplicada, EETAC, Universitat Politècnica de Catalunya BarcelonaTech, 08860 Castelldefels, Spain
cDepartment of Mechanical Engineering, University of South Florida, Tampa, FL 33620, USA
First published on 20th March 2014
Abstract
The catalytic effects of CoFe2O4 nanoparticles on the hydrogen storage properties of LiAlH4 prepared by ball milling were investigated. The onset desorption temperature of the LiAlH4 + 2 mol% CoFe2O4 sample is 65 °C, which is 90 °C lower that of the as-received LiAlH4, with approximately 7.2 wt% hydrogen released at 250 °C. The isothermal desorption results show that for the 2 mol% CoFe2O4 doped sample dehydrogenated at 120 °C, 6.8 wt% of hydrogen can be released within 160 min, which is 6.1 wt% higher than that of the as-received LiAlH4 under the same conditions. Through the differential scanning calorimetry (DSC) and the Kissinger desorption kinetics analyses, the apparent activation energy, Ea, of the 2 mol% CoFe2O4 doped sample is calculated as 52.4 kJ mol−1 H2 and 86.5 kJ mol−1 H2 for the first two decomposition processes. This is 42.4 kJ mol−1 H2 and 86.1 kJ mol−1 H2 lower compared with the pristine LiAlH4, respectively, indicating considerably improved dehydrogenation kinetics by doping the CoFe2O4 catalyst in the LiAlH4 matrix. From the Fourier transform infrared spectroscopy (FTIR) and X-ray diffraction (XRD) analyses, a series of finely dispersed Fe and Co species with a range of valence states, produced from the reactions between LiAlH4 and CoFe2O4, play a synergistic role in remarkably improving LiAlH4 dehydrogenation properties. The rehydrogenation properties of the LiAlH4 + 2 mol% CoFe2O4 sample have also been investigated at 140 °C under 6.5 MPa pressure held for 2.5 h.
1. Introduction
As a renewable energy source, hydrogen can be produced from water and biomass without any greenhouse gas emissions. Thus, hydrogen attracts considerable attention from research aiming to solve the fossil fuel depletion problem accompanied by the global environmental issues.1–3 The prerequisite for widespread hydrogen use as an energy carrier is the development of advanced hydrogen storage materials for safely storing it at high gravimetric and volumetric densities.4–6Among numerous possible hydrogen storage materials, lithium aluminum hydride7–10 (LiAlH4) is a promising candidate due to its relatively large theoretical hydrogen storage capacity and high potential reversible hydrogenation capability. Theoretically, LiAlH4 can desorb 10.5 wt% hydrogen upon heating to 420 °C, which make it an ideal hydrogen storage material to meet the U.S. Department of Energy 2015 targets for a viable hydrogen storage system11 with gravimetric density ≥5.5 wt% and volumetric density ≥40 g L−1. Upon heating, LiAlH4 would gradually release hydrogen, according to the following three steps.12 The first reaction step (R1) occurs in the 150–175 °C temperature range and releases 5.3 wt% hydrogen:
| 3LiAlH4 → Li3AlH6 + 2Al + 3H2 | (1) |
Then the second reaction step (R2) occurs between 180 °C and 220 °C, releasing 2.6 wt% hydrogen:
| Li3AlH6 + 2Al → 3LiH + 3Al + 3/2H2 | (2) |
The third reaction step (R3) starts to release 2.6 wt% hydrogen above 400 °C:
| 3LiH + 3Al → 3LiAl + 3/2H2 | (3) |
Thus, the dehydrogenation properties of LiAlH4 are generally analyzed for the first two decomposition reactions due to the high onset and decomposition temperatures, and the low desorbed hydrogen content of the reaction R3 from the practical applications perspective.13–16
Since Bogdanovic et al.17 conducted the seminal work in improving the hydrogen storage performance of NaAlH4 by doping TiCl3, extensive efforts have been devoted to ameliorate the re/dehydrogenation properties of LiAlH4 by adding various catalysts to lower its onset dehydrogenation temperature and increase its dehydrogenation kinetics. To date, the documented catalysts for LiAlH4 can be classified as: (1) pure metals;1,18–26 (2) carbon-containing species;21,27–31 (3) metal halides;13,19,21,32–42 (4) alloys;18,20 (5) metal oxides14–16,43,44 and (6) other compounds.45–49 To our knowledge, a partial reversibility can be realized through doping LiAlH4 with various catalysts.28,35,44,50 However, the rehydrogenation property was not ideal. From the practical applications perspective, solid-state materials (LiAlH4, NaAlH4, MgH2, etc.) do have the potential to outperform physical methods of storage (cryostorage or high-pressure technologies) through comprehensively considering the safety, environment friendless and cost, which has been reported in many review papers.51,52 However, it is crucial to find an advanced catalyst, which could not only significantly improve the dehydrogenation, but also rehydrogenation performance of LiAlH4. Recently the authors have observed the superior effects of Fe2O3 and Co2O3 nanoparticles on promoting the dehydrogenation properties of LiAlH4, however, nano-sized Fe2O3 and Co2O3 failed to produce any reversibility for LiAlH4.15 Herein, it is reasonable to speculate that Co ferrite shows a great potential as the catalyst to advance hydrogen storage performance of LiAlH4.
In this work, the catalytic efficiency of CoFe2O4 nanoparticles on the dehydrogenation and reversible hydrogenation properties of LiAlH4 was evaluated by utilizing a pressure-composition-temperature (PCT) apparatus and differential scanning calorimetry (DSC). The catalytic mechanism of CoFe2O4 nanoparticles was demonstrated by analyzing the results of the Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD) and scanning electronic microscopy (SEM). The comparison of the catalytic effects of CoFe2O4, Fe2O3 and Co2O3 catalysts for LiAlH4 is also presented in this work.
2. Experimental
2.1. Sample preparation
LiAlH4 (≥95% pure) was purchased from the Sigma Aldrich Co., and CoFe2O4 (≥99.99% pure, 20 nm) was prepared by using the sol–gel method. The details of the preparation procedure are given in the previous report.53 All handling of the samples was conducted in a glove box (Mikrouna Co., China) under high-purity argon atmosphere (H2O: <10 ppm; O2: <10 ppm) in order to minimize oxidation and humidity. About 1.5 g of LiAlH4 was mixed with various mole fractions of CoFe2O4 nanopowder, and then the mixture was loaded into a stainless steel grinding vial (5 cm in diameter, quenching). After that, ZrO2 balls (Mohs hardness ≥ 7.5) were added with a ball-to-powder weight ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in the glove box. Finally, the grinding vial with the mixed sample was ball milled for 30 min by using a high energy Spex mill (QM-3B) at a milling rate of 1200 rpm. In order to prevent excess heating and the surface fatigue wear of ball-milling materials, the grinding vial was cooled down for 5 min after milling every 10 min.
:1 in the glove box. Finally, the grinding vial with the mixed sample was ball milled for 30 min by using a high energy Spex mill (QM-3B) at a milling rate of 1200 rpm. In order to prevent excess heating and the surface fatigue wear of ball-milling materials, the grinding vial was cooled down for 5 min after milling every 10 min.
2.2. Characterization
The hydrogen storage performance of the as-received and doped LiAlH4 samples was measured by using a Sieverts-type PCT apparatus (Beijing Nonferrous Metal Research Institute, China). The PCT equipment can be heated up to 600 °C with a maximum hydrogen pressure of 10 MPa. To measure the dehydrogenation properties, 0.3 g sample was loaded into a stainless steel vessel and then heated to 250 °C at 5 °C min−1 heating rate under 0.1 atm pressure. For the rehydrogenation measurements, the samples that completed the first dehydrogenation were directly reheated at 150 °C under 6.5 MPa for 3 h. The de/rehydrogenation amount for all samples was calculated from the pressure changes, and then the values were converted for pure LiAlH4 with the elimination of various impurities, the detailed calculation formula is as follows,| mat-wt% = H2 mass/[mass (storage material) + mass (catalyst) + H2 mass] | (4) |
In order to investigate the decomposing behavior and calculate the activation energy of both as-received and doped LiAlH4 samples, DSC measurements were conducted by using NETZSCH STA 449C under a flow of 50 mL min−1 high-purity Ar. Typically, about 5 mg of sample was sealed into a 50 mL alumina crucible in the glove box, and then was heated at different heating rates (6 °C min−1, 9 °C min−1, and 12 °C min−1) from 35 °C to 300 °C, respectively.
The morphology of the as-received and 2 mol% CoFe2O4-doped samples were observed by SEM (ZEISS EVO 18, Germany) equipped with the energy dispersive spectroscopy (EDS) detector. Prior to the SEM observations, the samples were prepared inside the glove box, and then transferred to the SEM chamber in order to prevent oxidation and moisture adsorption.
FTIR analysis of the as-received and doped LiAlH4 samples after ball milling was carried out by using Bruker Vector 22 FTIR spectrometer. The FTIR spectra were recorded between 2000 cm−1 and 750 cm−1 with a spectral resolution of 4 cm−1.
Phase structure characteristics of the as-milled and de/rehydrogenated samples were detected by XRD (MXP21VAHF X-ray diffractometer with CuKα radiation, 40 kV, 200 mA) at room temperature. The X-ray intensity was tested over the 2θ angle ranged from 10° to 90° with a scanning velocity of 0.02° per second.
3. Results and discussion
Fig. 1 displays the non-isothermal desorption curves of the as-received LiAlH4, as-milled LiAlH4, and LiAlH4 doped with 1 mol%, 2 mol%, 3 mol%, and 5 mol% CoFe2O4 nanoparticles, heated from 25 °C to 250 °C at a heating rate of 5 °C min−1. As seen in Fig. 1, the as-received LiAlH4 sample started to release hydrogen at around 155 °C and about 5.0 wt% hydrogen desorbed during the first dehydrogenation step. With increasing temperature, the as-received LiAlH4 sample entered into the second dehydrogenation step from 200 °C and about 2.5 wt% hydrogen was released at the second dehydrogenation stage. Thus, the total hydrogen release capacity of 7.5 wt% could be obtained when the as-received LiAlH4 was heated to 250 °C. For the as-milled LiAlH4 sample, the onset dehydrogenation temperature in the first two dehydrogenation steps deceased by about 21 °C, compared with the as-received LiAlH4, mainly attributed to the surface activation, introduced to the LiAlH4 matrix by mechanical milling.13–16,24,28,38,39,42,43 Compared with the LiAlH4 samples without any catalysts doping, the onset desorption temperature of LiAlH4 doped with CoFe2O4 nanoparticles exhibited a remarkable reduction, not only for the first, but also for the second dehydrogenation step. When 1 mol% CoFe2O4 nanopowder was added to the LiAlH4 matrix, the onset dehydrogenation temperature decreased by 75 °C for the first stage and 40 °C for the second stage, compared with the as-received LiAlH4. The 1 mol% doped sample released 7.4 wt% hydrogen at the first two dehydrogenation steps. By further increasing the content of the CoFe2O4 nanoparticles to 2 mol%, the LiAlH4 + 2 mol% CoFe2O4 sample started to release hydrogen at 65 °C and 130 °C for the first two dehydrogenation steps, which decreased by 90 °C and 70 °C, compared with the as-received LiAlH4, respectively. Overall, 7.2 wt% hydrogen was released for the 2 mol% doped sample. For the hydrogen release content of 1 mol% and 2 mol% doped samples, they are close to the theoretical hydrogen release content of pristine LiAlH4 (7.5 wt% H2). For the 3 mol% CoFe2O4 doped sample, the onset dehydrogenation temperature further decreased to 61 °C for the first dehydrogenation step, while only 5.5 wt% hydrogen was released during the first two dehydrogenation processes, indicating a drastic reduction in the released hydrogen capacity after doping an excess amount of CoFe2O4 nanoparticles. A similar phenomenon was also proposed in previous reports.13,14,28,35,37,43,47 However, when 5 mol% of CoFe2O4 were added, the LiAlH4 doped sample started to dehydrogenate at 100 °C, which is much higher than the other contents CoFe2O4-doped samples. Meanwhile, the desorption hydrogen content dropped sharply to 3.2 wt% for the first two dehydrogenation steps, which only accounts for 41.7% of the total hydrogen release for pure LiAlH4. The excessive decrease in the amount of hydrogen release for the LiAlH4 + 5 mol% CoFe2O4 samples contributes to the excessive catalytic effect, leading to the complete decomposition of LiAlH4 during the high-energy ball-milling process. In the meanwhile, the dehydrogenation process conducted during the heating and desorption process was the second desorption stage only. Fig. 2 shows hydrogen released from LiAlH4 doped with different amounts of CoFe2O4, Fe2O3 and Co2O3 catalysts, which is nearly close to the theoretical hydrogen release content of the pristine LiAlH4. However, when the content of every catalyst is higher than a certain value, the amount of hydrogen released sharply decreases. For the CoFe2O4 doped LiAlH4 sample, its hydrogen released amount declined quickly when more than 2 mol% CoFe2O4 nanoparticles were added. However, as for the Fe2O3 and Co2O3 doped LiAlH4 samples, their hydrogen release content decreases rapidly when the Fe2O3 and Co2O3 nanoparticles content was more than 5 mol%. CoFe2O4 has a stronger catalytic effect on the dehydrogenation properties of LiAlH4, compared with Fe2O3 and Co2O3. The LiAlH4 + 2 mol% CoFe2O4 sample exhibits optimal dehydrogenation performance, based on the onset dehydrogenation temperature and hydrogen desorption capacity, and would be utilized to analyze the catalytic effect and the mechanism of the CoFe2O4 nanoparticles in the following tests. | ||
| Fig. 1 Thermal desorption profiles of the as-received LiAlH4, as-milled LiAlH4, and LiAlH4 doped with 1, 2, 3, and 5 mol% CoFe2O4 nanoparticles. The samples are heated to 250 °C at 5 °C min−1 heating rate. | ||
 | ||
| Fig. 2 Hydrogen released from LiAlH4 doped with different catalysts in the 25–250 °C temperature range. | ||
Fig. 3 shows the isothermal dehydrogenation behavior of the as-received LiAlH4 at 120 °C and the LiAlH4 + 2 mol% CoFe2O4 at 90 °C, 120 °C and 150 °C, respectively. From the curve (a) in Fig. 3, only 0.7 wt% of hydrogen could be detected within 180 min, indicating a perishing desorption kinetics of pristine LiAlH4 at 120 °C. However, the dehydrogenation kinetics of LiAlH4 was significantly enhanced after doping Co ferrite nanopowder. When heated at 90 °C (Fig. 3b), the CoFe2O4-doped sample could release 5.1 wt% hydrogen within 160 min, suggesting the first dehydrogenation step completion for LiAlH4. Furthermore, the 2 mol% doped sample released 6.8 wt% of hydrogen within 160 min at 120 °C (Fig. 3c), which is 6.1 wt% higher compared with the as-received LiAlH4 for the same heating temperature and time. When further increasing temperature up to 150 °C, only 55 min were required to complete the first two dehydrogenation steps for the LiAlH4 doped with 2 mol% CoFe2O4, as seen in Fig. 2d. Thus it is reasonable to conclude that CoFe2O4 exhibits superior catalytic performance and significantly improves the dehydrogenation kinetics of LiAlH4, which makes it quite attractive for the PEM fuel cell applications.
 | ||
| Fig. 3 Isothermal dehydrogenation kinetics of (a) as-received LiAlH4 at 120 °C, and LiAlH4 + 2 mol% CoFe2O4 at: (b) 90 °C, (c) 120 °C, and (d) 150 °C. (I) represents the first dehydrogenation step, and (II) presses the second dehydrogenation step. | ||
To further reflect the CoFe2O4 nanoparticles excellent catalytic effect of improving the LiAlH4 isothermal dehydrogenation kinetics and test the practical operating temperature of the PEM fuel cells, Fig. 4 shows isothermal dehydrogenation kinetics of LiAlH4 doped with CoFe2O4, Fe2O3 and Co2O3 heated at 90 °C. As seen in Fig. 4, the Co2O3 and Fe2O3 doped samples release 4.0 wt% and 4.4 wt% H2 in 180 min at 90 °C, while the CoFe2O4 doped sample could release 5.1 wt% H2 within 160 min, indicating that CoFe2O4 is superior to Fe2O3 and Co2O3 in improving the dehydrogenation kinetics of LiAlH4. This is in good agreement with the hydrogen released amount results of LiAlH4 doped with these three catalysts (Fig. 2).
 | ||
| Fig. 4 Isothermal dehydrogenation kinetics of LiAlH4 doped with 2 mol% CoFe2O4, 5 mol% Fe2O3 and 5 mol% Co2O3 heated at 90 °C. | ||
In order to further analyze the dehydrogenation steps of the CoFe2O4 doped samples in terms of the exo/endothermic characteristics and to acquire activation energy (Ea) for each dehydrogenation step according to the Kissinger method, Fig. 5 displays the DSC curves of the as-received LiAlH4 (6 °C min−1) and 2 mol% CoFe2O4 doped LiAlH4 (6 °C min−1, 9 °C min−1 and 12 °C min−1) within the 35–300 °C temperature range, respectively. The as-received LiAlH4 DSC curve contains four characteristic peaks in the first two dehydrogenation steps (two exothermic and two endothermic peaks). These four thermal characteristic peaks correspond to the interaction of LiAlH4 with surface hydroxyl impurities at 154.9 °C, melting of LiAlH4 at 166.4 °C,54 decomposition of liquid LiAlH4 (R1) at 184.5 °C and decomposition of Li3AlH6 at 240 °C (R2).32 However, the DSC curve of the CoFe2O4 doped LiAlH4 sample has only two characteristic peaks measured at different heating rates. When heated at a heating rate of 6 °C min−1, the exothermic peak of the doped sample appeared at about 131 °C. Thus the first exothermic peak is attributed to the decomposition of the solid state LiAlH4, since the CoFe2O4 doped LiAlH4 started to decompose prior to its melting. Then the endothermic peak emerged at 205 °C, corresponding to the dehydrogenation step of Li3AlH6. Furthermore, the characteristic temperatures of these two endothermic peaks gradually rise with the increasing heating rate, suggesting that the doped sample has more time to relax at any given temperature and thus the decomposition occurs sooner at a lower temperature when heated at the relatively lower rate. A similar phenomenon is also reported in the DSC results of LiAH4 doped with various catalysts.13–16,18,32,38,40,43,44 Therefore, the dehydrogenation properties of LiAlH4 were significantly improved by adding CoFe2O4 nanoparticles, reflecting the remarkable reduction on the characteristic peak temperature of LiAH4.
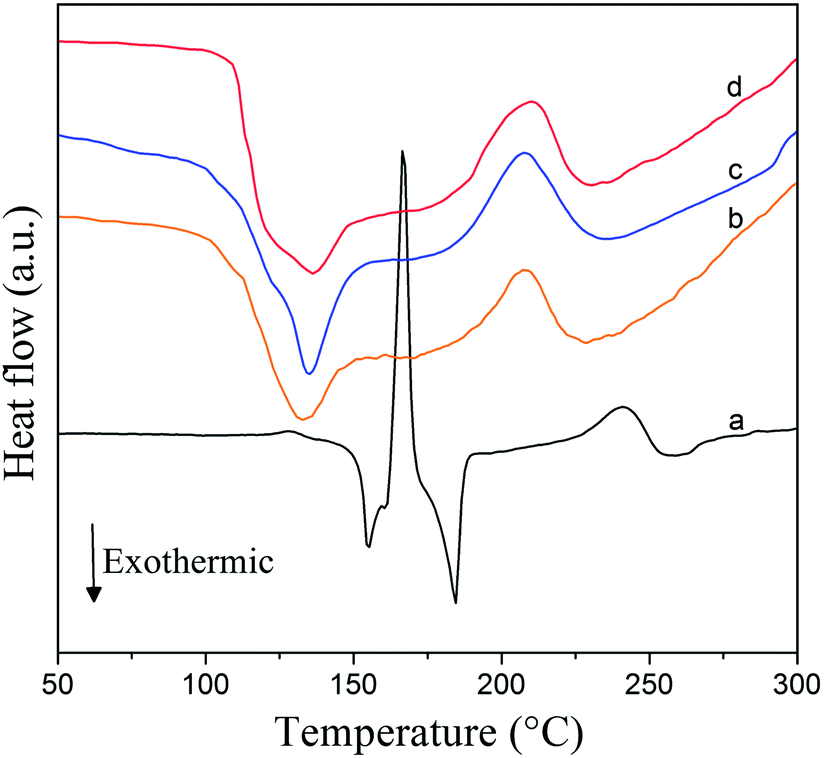 | ||
| Fig. 5 DSC curves of (a) as-received LiAlH4, LiAlH4 + 2 mol% CoFe2O4 in the 35–300 °C temperature range and the heating rate of: (b) 6 °C min−1, (c) 9 °C min−1, and (d) 12 °C min−1. | ||
In order to analyze the catalytic mechanism of CoFe2O4 nanoparticles on the dehydrogenation properties of LiAlH4, the apparent activation energy (Ea) of the as-received LiAlH4 and the CoFe2O4-doped LiAlH4 sample for the first two decomposition steps were calculated by using the Kissinger method,55
 | (5) |
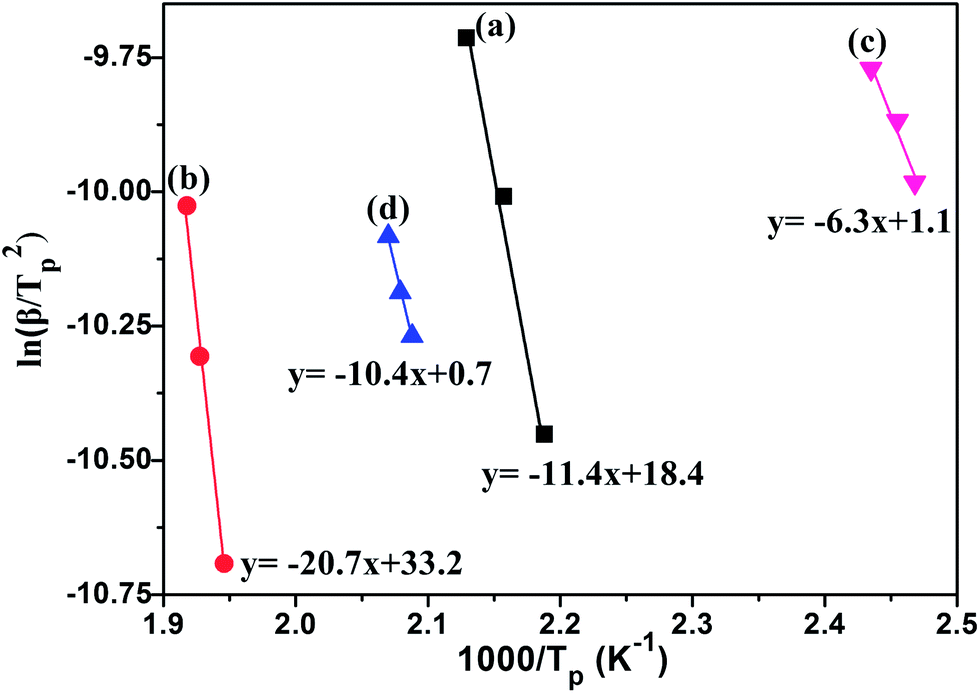 | ||
| Fig. 6 Kissinger plots for the as-received LiAlH4: (a) the first step and (b) the second step and LiAlH4 doped with 7 mol% CoFe2O4: (c) the first step and (d) the second step. | ||
In order to compare the change of morphology of the powder samples before/after ball milling and show the distribution of constitution elements of catalyst around the LiAlH4 matrix, Fig. 7 presents the SEM images of the as-received LiAlH4, ball-milled LiAlH4 and the 2 mol% doped LiAlH4 coupled with the elemental maps. As seen in Fig. 7a, the as-received LiAlH4 sample consists of large irregular polyhedron particles, up to 40 μm in size. However, in Fig. 7b, the morphology of the as-milled LiAlH4 became as amounts of regular globular particles with diameter ranging from 3 and 10 μm, reflecting a significant decrease in the particle size of LiAlH4 after ball milling. Fig. 7(c–g) display the SEM images and the corresponding elemental maps of the LiAlH4 + 2 mol% CoFe2O4 sample after mechanical ball-milling for 30 min. Microscopically, the grains of the LiAlH4 + 2 mol% CoFe2O4 sample are fine but inhomogeneous, and the original particles were broken into smaller particles with the average size of about 6 μm by mechanical ball-milling. The tiny particles have a tendency to assemble and form stepped structures. As seen in Fig. 7(d–g), the elemental maps of constituent elements Al, O, Fe, and Co show uniform distribution of these species in the mixture, indicating that the catalyst of CoFe2O4 nanopowder could be well mixed with LiAlH4 matrix through high energy ball milling. There is an existing good contact between the CoFe2O4 catalyst and the LiAlH4 particles, resulting in the significantly enhanced dehydrogenation kinetics of LiAlH4. Nevertheless, through comparison the elemental map O with that of other constituent elements of CoFe2O4 catalyst, it is worth to note that the elemental map of O has more distribution than that of Fe and Co, which is mainly caused by the oxidation during the specimen preparation process and oxygen element introduced from the conducting resin. Therefore, the high density surface defects and well dispersed catalyst introduce a larger amount of reaction nucleation sites the and hydrogen diffusion channels around the LiAlH4 matrix for the dehydrogenation process, which results in the surface activation and obviously improved dehydrogenation properties of LiAlH4.
 | ||
| Fig. 7 SEM micrographs of (a) as-received LiAlH4 and (b) LiAlH4 + 2 mol% CoFe2O4 after ball-milling. (c) SEM micrograph with (d)–(g) corresponding elemental maps of the 2 mol% CoFe2O4-doped sample. | ||
IR spectra of the as-received LiAlH4, as-milled LiAlH4 and LiAlH4 doped with 1 mol%, 2 mol%, 3 mol% and 5 mol% CoFe2O4 samples after ball milling are compared in Fig. 8. According to ref. 14, 24, 28, 40, 44 and 46, the active infrared vibrations of the Al–H bond for LiAlH4 distribute at two regions, corresponding to 1600–1800 cm−1 for the Al–H stretching modes and 800–900 cm−1 for the Li–Al–H bending modes. While the active infrared vibrations for Li3AlH6 exhibit the Al–H stretching modes in the 1500–1400 cm−1 region.14–16,28,44,56 For the CoFe2O4 doped LiAlH4 samples shown in Fig. 8 (curves c–f), their active infrared vibration of the Al–H stretching modes appear at 1473 cm−1, suggesting the existence of Li3AlH6 in the doped sample after ball milling. However, no Al–H bond peak of Li3AlH6 is found at the same position in the IR spectra of the as-received and as-milled LiAlH4 (Fig. 8, curves a and b). The absorption intensity of the Li3AlH6 peak gradually strengthen with increasing CoFe2O4 catalyst content, which indicates that the content of Li3AlH6 continuously increases resulting from the decomposition proportion of LiAlH4 raise with more CoFe2O4 catalyst. It is worth to note that the LiAlH4 IR absorption peak cannot be observed when adding 5 mol% CoFe2O4 nanoparticles into the LiAlH4 matrix, resulting from the 5 mol% CoFe2O4 doped sample complete decomposition and Li3AlH6 formation during the ball milling process. This phenomenon can be confirmed by the nonisothermal dehydrogenation performance of the 5 mol% doped LiAlH4 (Fig. 1). Based on the comprehensive IR spectra analysis, it is concluded that the CoFe2O4-doped LiAlH4 decomposition reaction occurs, forming the Li3AlH6 phase during the ball-milling process. The decomposition reaction of LiAlH4 gradually intensifies with the increasing CoFe2O4 amount, and the details of the decomposition byproducts would be determined by the following XRD measurements.
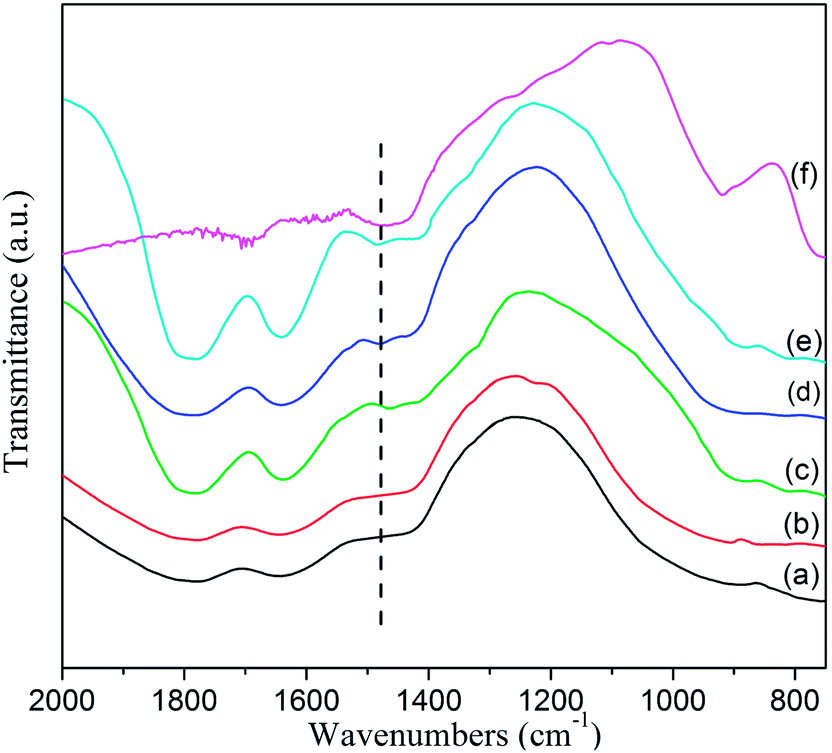 | ||
| Fig. 8 FTIR spectra of (a) as-received LiAlH4, (b) as-milled LiAlH4 and (c) 1 mol%, (d) 2 mol%, (e) 3 mol% and (f) 5 mol% CoFe2O4 doped LiAlH4 after ball milling. | ||
The above measurements confirm that some specific stoichiometric reactions between LiAlH4 and CoFe2O4 occur during the ball-milling process. To clarify the phase transforms between LiAlH4 and CoFe2O4 during the ball-milling process, Fig. 9 presents the XRD patterns of the as-milled LiAlH4 and LiAlH4 doped with 2 mol%, 3 mol% and 5 mol% CoFe2O4 after the ball milling process. In the XRD spectra of the as-milled LiAlH4 all diffraction peaks correspond to the LiAlH4 phase, without any additional decomposition products, suggesting that pure LiAlH4 remains rather stable during the ball milling process.13–16,18,40,43,57,58 This point can also be proven by the non-isothermal dehydrogenation properties of the as-received and as-milled LiAlH4 (Fig. 1), and the FTIR spectra of the as-milled LiAlH4 (Fig. 8). However, compared with the as-milled LiAlH4, the XRD patterns of the CoFe2O4 doped LiAlH4 samples do not appear as just physical mixtures of LiAlH4 and Co ferrite, which is in a good agreement with the FTIR results (Fig. 8). Adding 2 mol% CoFe2O4 nanoparticles into the LiAlH4 matrix by mechanical milling causes weak diffraction peaks of microcrystalline aluminum and Li3AlH6 to appear in the XRD pattern. Meanwhile, the diffraction peaks of LiFeO2 are observed at 41.3°, 44.7° and 34.8°, and the diffraction peaks at 31.3°and 44.8° correspond to AlCo, while the diffraction peaks of Fe3O4 are at 44.8°, and 65.1°. However, the CoFe2O4 phase could not be detected in the XRD patterns for the doped samples after the ball milling, which demonstrates that the reaction between LiAlH4 and CoFe2O4 occurred during the ball-milling process. A similar phenomenon also appears in LiAlH4 doped with other documented nanosized catalysts: MnFe2O4,14 Fe2O3,15 NiFe2O416 and Nb2O5,44 in which a complete reaction occurs between LiAlH4 and the catalyst precursor, and subsequently the reaction products act as real catalysts for the succeeding decomposition of LiAlH4. With increasing the Co ferrite content up to 3 mol%, the diffraction intensity of the decomposition products of Al, Li3AlH6, LiFeO2 and Fe3O4 is gradually enhanced. The diffraction intensity of LiAlH4 conspicuously declines, compared with that of 2 mol% doped LiAlH4 sample, signifying that LiAlH4 reacts with CoFe2O4, resulting in more LiAlH4 decomposition during the ball milling process. Surprisingly, the diffraction peaks of LiAlH4 cannot be observed for the 5 mol% doped sample, and all diffraction peaks correspond to the decomposition products, including LiFeO2, Fe3O4, AlCo, Al and Li3AlH6, as seen in Fig. 9. This can be explained by the reaction between LiAlH4 and CoFe2O4, leading to the complete decomposition of LiAlH4 doped with CoFe2O4 during the ball milling process, causing the LiAlH4 phase disappearance in the 5 mol% CoFe2O4-doped sample. In addition, the nano-sized CoFe2O4 phase cannot be detected in the XRD patterns of all doped samples, mainly because of the complete reaction between LiAlH4 and CoFe2O4, forming LiFeO2, AlCo, Al and Li3AlH6 phases. In the literature, a similar phenomenon has been reported, where NbF5−,13 MnFe2O4−,14 NiCl2−,37 TiF3−,38 and TiO2−,43 as additives for LiAlH4 also could not be detected after high energy ball-milling.
 | ||
| Fig. 9 XRD patterns for the as-milled LiAlH4 and LiAlH4 + 2 mol%, 3 mol% and 5 mol% CoFe2O4 after ball milling. | ||
Fig. 10 displays the XRD patterns of the as-milled LiAlH4 and 2 mol%, 3 mol% and 5 mol% CoFe2O4-doped LiAlH4 after dehydrogenation at 250 °C. The XRD spectra of dehydrogenated as-milled LiAlH4 only consists of Al and LiH phases as the dehydrogenation products, demonstrating that the first two dehydrogenation steps of LiAlH4 have completed upon heating to 250 °C. In contrast, the XRD patterns of the doped samples show the dehydrogenation products containing not only Al and LiH phases, but also LiFeO2, LiAlO2, Fe0.98O and Al0.52Co0.48 phases, which is quite different compared with the dehydrogenation products of the as-milled counterpart samples. Moreover, the diffraction peaks of LiFeO2, LiAlO2, Fe0.98O and Al0.52Co0.48 phases gradually strengthen with the increasing CoFe2O4 amount. With respect to the significantly improved dehydrogenation performance of LiAlH4 by doping CoFe2O4 nanoparticles, in situ formed reaction products may act as the catalyst for the first two dehydrogenation steps of LiAlH4. Meanwhile, the reactions occurring during the dehydrogenation processes could facilitate the dehydrogenation dynamics of LiAlH4. These favorable factors together provide a synergetic contribution to the significantly improved dehydrogenation properties of LiAlH4.
 | ||
| Fig. 10 XRD patterns of the as-milled LiAlH4 and LiAlH4 + 2 mol%, 3 mol% and 5 mol% CoFe2O4 after dehydrogenation at 250 °C. | ||
The above experimental results demonstrate that the CoFe2O4 nanopowder plays an important role in improving the dehydrogenation properties of Li alanate. The reasons leading to the significantly improved dehydrogenation properties, acquired in this work for the CoFe2O4-doped samples, could be summarized as follows: first, previous studies have revealed that the reaction thermodynamics could be affected by reducing the grain size.59 The smaller particle size and a large number of created surface defects can introduce more reaction nucleation sites and hydrogen diffusion channels for the dehydrogenation process of LiAlH4. Second, CoFe2O4 reacts with LiAlH4 during the ball-milling process by forming a ternary Li–Fe oxide (LiFeO2), Al–Co compound (AlCo) and Fe oxide (Fe3O4) species, suggesting that Co ferrite can transform into other new Co- and Fe-containing phases by increasing the high local temperature (demonstrated in ESI†) during the ball milling process. After dehydrogenation, the LiFeO2, LiAlO2, Fe0.98O and Al0.52Co0.48 phases as the dehydrogenation products appear in the XRD patterns, and the diffraction intensity of these products gradually increases with further CoFe2O4 additive amount. These finely dispersed reaction products serve as the active sites for nucleation and growth of the dehydrogenation products, and the diffusion length of the reaction ions is largely shortened. Third, series of reactions between LiAlH4 and CoFe2O4 occur by forming a ternary Li–Fe oxide, Fe oxide and Al–Co phases with a reduced valence state during heating. Thus, it is expected that these reactions could alter the reaction thermodynamics by lowering the enthalpy of the dehydrogenation reaction.44 It is reasonable to conclude that the refinement of the LiAlH4 powder combined with the reactions between LiAlH4 and CoFe2O4 together contribute to the significantly improved dehydrogenation kinetics of LiAlH4.
In order to comprehensively consider the catalytic effect of nano-sized CoFe2O4 for LiAlH4, Fig. 11 shows the rehydrogenation results of the 2 mol% doped sample at 140 °C under 6.5 MPa pressure, followed by the subsequent desorption at 250 °C. After complete dehydrogenation during the first two reactions heated up to 250 °C, the sample was rehydrogenated at 140 °C under 6.5 MPa pressure. It is obvious that the rehydrogenation properties of the CoFe2O4 doped sample reach 0.15 wt% H2 resorbed for the given conditions. Meanwhile, in order to confirm the rehydrogenation effect, Fig. 11 also provides the XRD pattern of the LiAlH4 + 2 mol% CoFe2O4 sample after resorbing hydrogen for the given conditions in 2.5 h. The XRD spectra of the rehydrogenated sample shows almost identical results with the dehydrogenated sample, except for the appearance of few Li3AlH6 peaks, indicating that the second decomposition reaction of LiAlH4 may be partially reversible by the catalytic effects of Co- and Fe-containing products. However, further study of hydrogen storage reversibility of the dehydrogenated LiAlH4 is still underway.
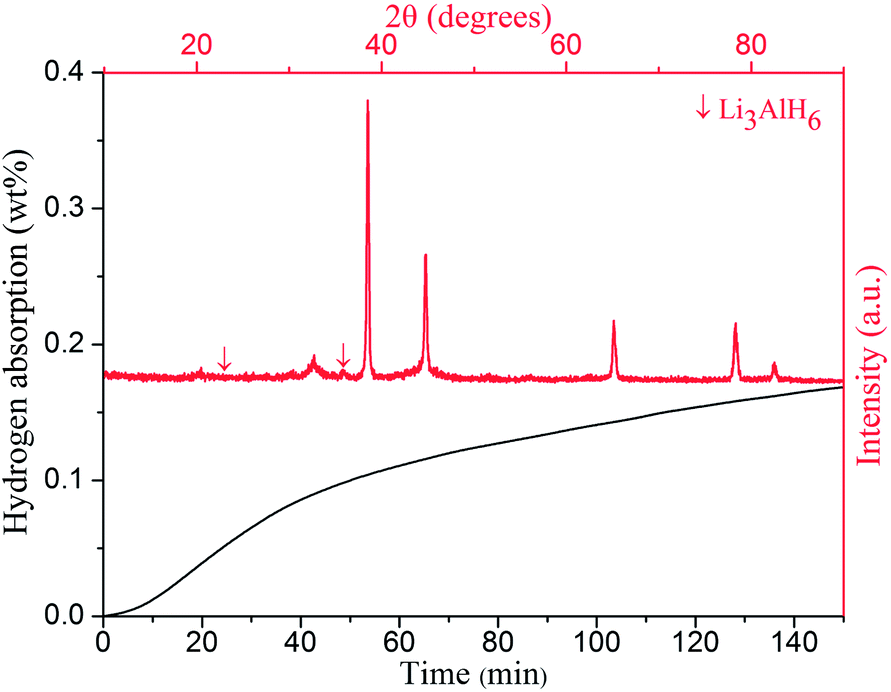 | ||
| Fig. 11 Rehydrogenation of LiAlH4 + 2 mol% CoFe2O4 sample and its corresponding XRD pattern after hydrogen resorbtion at 140 °C under 6.5 MPa H2 for 2.5 h. | ||
4. Conclusions
In summary, the dehydrogenation properties of LiAlH4 catalyzed by CoFe2O4 nanoparticles have been substantially improved compared with pure Li alanate powder. The onset desorption temperature of the 2 mol% CoFe2O4 doped LiAlH4 sample is 65 °C, resulting in 90 °C decrease, compared with the as-received LiAlH4. The rehydrogenation properties of the 2 mol% CoFe2O4 doped LiAlH4 are inferior for the tested conditions, with 0.15 wt% H2 resorbtion. The isothermal dehydriding kinetics shows that the LiAlH4 + 2 mol% CoFe2O4 sample can release 6.8 wt% of hydrogen in 160 min under 0.1 MPa pressure, which is 6.1 wt% higher than that of the pristine LiAlH4 under the same conditions (time, temperature and pressure). Furthermore, through the differential scanning calorimetry and the Kissinger desorption kinetics analyses, the apparent activation energy, Ea, of the 2 mol% CoFe2O4 doped sample are calculated to be 52.4 kJ mol−1 H2 and 86.5 kJ mol−1 H2 for the first two decomposition reactions, which are 42.4 kJ mol−1 H2 and 86.1 kJ mol−1 H2 lower than those of the pristine LiAlH4, respectively. Based on the FTIR and XRD analyses of the doped samples, a series of reactions occurred between LiAlH4 and CoFe2O4 during the ball-milling process, forming Al, Li3AlH6, LiFeO2, Fe3O4, and Fe3O4 as decomposition products. These reactions proceeded upon heating, and the LiFeO2, LiAlO2, Fe0.98O and Al0.52Co0.48 phases appeared. These in situ formed decomposition products, coupled with the reactions, play a synergistic role in remarkably improving dehydrogenation properties of LiAlH4. From the conducted experiments it is reasonable to conclude that CoFe2O4 nanoparticles play a critical role in the significantly improved LiAlH4 dehydrogenation performance.Acknowledgements
The authors thank the National High-Tech R&D Program (863 Program) of China (2006AA05Z132) for financial support of this research. Fuqiang Zhai thanks the China Scholarship Council (CSC) for the scholarship.References
- L. G. Li, Q. F. Gu, Z. W. Tang, X. W. Chen, Y. B. Tan, Q. Li and X. B. Yu, J. Mater. Chem. A, 2013, 1, 12263–12269 CAS.
- M. Ismail, Y. Zhao, X. B. Yu and S. X. Dou, RSC Adv., 2011, 1, 408–414 RSC.
- R. Din, X. H. Qu, P. Li, L. Zhang, M. Ahmad, M. Z. Iqbal, M. Y. Rafique and M. H. Farooq, RSC Adv., 2012, 2, 4891–4903 RSC.
- P. Sridechprasat, L. Phuirot, P. Rangsunvigit, B. Kitiyanan and S. Kulprathipanja, Energies, 2012, 5, 3691–3700 CrossRef CAS PubMed.
- F. Y. Cheng, Z. L. Tao, J. Liang and J. Chen, Chem. Commun., 2012, 48, 7334–7343 RSC.
- H. Reardon, J. M. Hanlon, R. W. Hughes, A. G. Jopek, T. K. Mandalac and D. H. Gregory, Energy Environ. Sci., 2012, 5, 5951–5979 CAS.
- A. Borgschulte, A. Jain, A. J. Ramirez-Cuesta, P. Martelli, A. Remhof, O. Friedrichs, R. Gremaud and A. Züttel, Faraday Discuss., 2011, 151, 213–230 RSC.
- J. C. Fallas, W. M. Chien, D. Chandra, V. K. Kamisetty, E. D. Emmons, A. M. Covington, R. Chellappa, S. A. Gramsch, R. J. Hemley and H. Hagemann, J. Phys. Chem. C, 2010, 114, 11991–11997 CAS.
- Y. B. Tan and X. B. Yu, RSC Adv., 2013, 3, 23879–23894 RSC.
- D. Lacina, L. Yang, I. Chopra, J. Muckerman, Y. Chabal and J. Graetz, Phys. Chem. Chem. Phys., 2012, 14, 6569–6576 RSC.
- http://www1.eere.energy.gov/hydrogenandfuelcells/storage/pdfs/targets_onboard_hydro_storage_explanation.pdf.
- A. Andreasen, T. Veggea and A. S. Pedersena, J. Solid State Chem., 2005, 178, 3672–3678 CrossRef CAS PubMed.
- M. Ismail, Y. Zhao, X. B. Yu and S. X. Dou, Int. J. Hydrogen Energy, 2010, 35, 2361–2367 CrossRef CAS PubMed.
- F. Q. Zhai, P. Li, A. Z. Sun, S. Wu, Q. Wan, W. N. Zhang, Y. L. Li, L. Q. Cui and X. H. Qu, J. Phys. Chem. C, 2012, 116, 11939–11945 CAS.
- Z. L. Li, P. Li, Q. Wan, F. Q. Zhai, Z. W. Liu, K. F. Zhao, L. Wang, S. Y. Lü, L. Zou, X. H. Qu and A. A. Volinsky, J. Phys. Chem. C, 2013, 117, 18343–18352 CAS.
- P. Li, Z. L. Li, Q. Wan, F. Q. Zhai, X. Q. Li, X. H. Qu and A. A. Volinsky, J. Phys. Chem. C, 2013, 117, 25917–25925 CAS.
- B. Bogdanovic and M. Schwickardi, J. Alloys Compd., 1997, 253, 1–9 CrossRef.
- V. P. Balema, V. K. Pecharsky and K. W. Dennis, J. Alloys Compd., 2000, 313, 69–74 CrossRef CAS.
- J. Chen, N. Kuriyama, Q. Xu, H. T. Takeshita and T. Sakai, J. Phys. Chem. B, 2001, 105, 11214–11220 CrossRef CAS.
- V. P. Balema, J. W. Wiench, K. W. Dennis, M. Pruski and V. K. Pecharsky, J. Alloys Compd., 2001, 329, 108–114 CrossRef CAS.
- M. Resan, M. D. Hampton, J. K. Lomness and D. K. Slattery, Int. J. Hydrogen Energy, 2005, 30, 1413–1416 CrossRef CAS PubMed.
- X. P. Zheng, P. Li, F. Q. An, G. Q. Wang and X. H. Qu, Rare Met. Mater. Eng., 2008, 37, 400–403 CrossRef CAS.
- N. Mehraj-ud-din, R. Sami-ullah, C. S. So, S. W. Hwang, A. R. Kim and K. S. Nahm, Int. J. Hydrogen Energy, 2009, 34, 8937–8943 CrossRef PubMed.
- R. A. Varin and L. Zbroniec, J. Alloys Compd., 2010, 506, 928–939 CrossRef CAS PubMed.
- H. W. Langmi, G. S. McGrady, X. F. Liu and C. M. Jensen, J. Phys. Chem. C, 2010, 114, 10666–10669 CAS.
- X. F. Liu, H. W. Langmi, S. D. Beattie, F. F. Azenwi, G. S. McGrady and C. M. Jensen, J. Am. Chem. Soc., 2011, 133, 15593–15597 CrossRef CAS PubMed.
- K. L. Hima, B. Viswanathan and M. S. Srinivasa, Int. J. Hydrogen Energy, 2008, 33, 366–373 CrossRef PubMed.
- R. Din, L. Zhang, P. Li and X. H. Qu, J. Alloys Compd., 2010, 508, 119–128 CrossRef PubMed.
- L. H. Kumar, C. V. Rao and B. Viswanathan, J. Mater. Chem. A, 2013, 1, 3355–3361 CAS.
- C. P. Hsu, D. H. Jiang, S. L. Lee, J. L. Horng, M. D. Gerb and J. K. Chang, Chem. Commun., 2013, 49, 8845–8847 RSC.
- W. C. Hsu, C. H. Yang and W. T. Tsai, Int. J. Hydrogen Energy, 2014, 39, 927–933 CrossRef CAS PubMed.
- A. Andreasen, J. Alloys Compd., 2006, 419, 40–44 CrossRef CAS PubMed.
- J. R. Ares Fernandez, K. F. Aguey-Zinsou, M. Elsaesser, X. Z. Ma, M. Dornheim, T. Klassen and R. Bormann, Int. J. Hydrogen Energy, 2007, 32, 1033–1040 CrossRef CAS PubMed.
- Y. Suttisawat, P. Rangsunvigit, B. Kitiyanana, N. Muangsinb and S. Kulprathipanjac, Int. J. Hydrogen Energy, 2007, 32, 1277–1285 CrossRef CAS PubMed.
- X. P. Zheng, P. Li, I. S. Humail, F. Q. An, G. Q. Wang and X. H. Qu, Int. J. Hydrogen Energy, 2007, 32, 4957–4960 CrossRef CAS PubMed.
- Y. Kojima, Y. Kawai, M. Matsumoto and T. Haga, J. Alloys Compd., 2008, 462, 275–278 CrossRef CAS PubMed.
- T. Sun, C. K. Huang, H. Wang, L. X. Sun and M. Zhu, Int. J. Hydrogen Energy, 2008, 33, 6216–6221 CrossRef CAS PubMed.
- S. S. Liu, L. X. Sun, Y. Zhang, F. Xu, J. Zhang, H. L. Chu, M. Q. Fan, T. Zhang, X. Y. Song and J. P. Grolier, Int. J. Hydrogen Energy, 2009, 34, 8079–8085 CrossRef CAS PubMed.
- R. A. Varin and L. Zbroniec, J. Alloys Compd., 2011, 509S, S736–S739 CrossRef PubMed.
- Z. B. Li, S. S. Liu, X. L. Si, J. Zhang, C. L. Jiao, S. Wang, S. Liu, Y. J. Zou, L. X. Sun and F. Xu, Int. J. Hydrogen Energy, 2012, 37, 3261–3267 CrossRef CAS PubMed.
- J. Fu, L. Röntzsch, T. Schmidt, M. Tegel, T. Weißgärber and B. Kieback, Int. J. Hydrogen Energy, 2012, 37, 13387–13392 CrossRef CAS PubMed.
- X. P. Zheng, J. J. Zheng, Q. H. Ma, S. L. Liu, X. Feng, X. B. Lin and G. Xiao, J. Alloys Compd., 2013, 551, 508–511 CrossRef CAS PubMed.
- M. Ismail, Y. Zhao, X. B. Yu, I. P. Nevirkovets and S. X. Dou, Int. J. Hydrogen Energy, 2011, 36, 8327–8334 CrossRef CAS PubMed.
- R. Din, X. H. Qu, P. Li, L. Zhang and M. Ahmad, J. Phys. Chem. C, 2011, 115, 13088–13099 Search PubMed.
- X. P. Zheng and S. L. Liu, J. Alloys Compd., 2009, 481, 761–763 CrossRef CAS PubMed.
- M. Ismail, Y. Zhao, X. B. Yu, A. Ranjbar and S. X. Dou, Int. J. Hydrogen Energy, 2011, 36, 3593–3599 CrossRef CAS PubMed.
- S. S. Liu, Z. B. Li, C. L. Jiao, X. L. Si, L. N. Yang, J. Zhang, H. Y. Zhou, F. L. Huang, Z. Gabelica, C. Schick, L. X. Sun and F. Xu, Int. J. Hydrogen Energy, 2013, 38, 2770–2777 CrossRef CAS PubMed.
- L. Li, F. Y. Qiu, Y. J. Wang, Y. N. Xu, C. H. An, G. Liu, L. F. Jiao and H. T. Yuan, Int. J. Hydrogen Energy, 2013, 38, 3695–3701 CrossRef CAS PubMed.
- L. Li, Y. N. Xu, Y. Wang, Y. J. Wang, F. Y. Qiu, C. H. An, L. F. Jiao and H. T. Yuan, Dalton Trans., 2014, 43, 1806–1813 RSC.
- X. F. Liu, S. D. Beattie, H. W. Langmi, G. S. McGrady and C. M. Jensen, Int. J. Hydrogen Energy, 2012, 37, 10215–10221 CrossRef CAS PubMed.
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- S. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS PubMed.
- M. George, S. S. Nair, K. A. Malini, P. A. Joy and M. R. Anantharaman, J. Phys. D: Appl. Phys., 2007, 40, 1593–1602 CrossRef CAS.
- M. McCarty, J. N. Maycock and V. R. P. Verneker, J. Phys. Chem., 1968, 72, 4009–4014 CrossRef CAS.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- S. S. Liu, Y. Zhang, L. X. Sun, J. Zhang, J. N. Zhao, F. Xu and F. L. Huang, Int. J. Hydrogen Energy, 2010, 35, 4554–4561 CrossRef CAS PubMed.
- R. A. Varin, L. Zbroniec, T. Czujko and Z. S. Wronski, Int. J. Hydrogen Energy, 2011, 36, 1167–1176 CrossRef CAS PubMed.
- P. B. Amama, J. T. Grant, P. J. Shamberger, A. A. Voevodin and T. S. Fisher, J. Phys. Chem. C, 2012, 116, 21886–21894 CAS.
- J. Lu, Y. J. Choi, Z. Z. Fang, H. Y. Sohn and E. Ronnebro, J. Am. Chem. Soc., 2009, 131, 15843–15852 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD pattern for the as-milled LiAlH4 doped with CoFe2O4 by using hand-milling method is provided in order to explain the fact that temperature is driving force for the reaction between LiAlH4 and CoFe2O4. See DOI: 10.1039/c4ra00841c |
| This journal is © The Royal Society of Chemistry 2014 |