Stereoselective inversion of γ-vinyl-γ-butyrolactone under palladium catalysis: application to the synthesis of (+)-exo- and (+)-endo-brevicomins†
Rodney A. Fernandes*,
Pullaiah Kattanguru and
Venkati Bethi
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400 076, Maharashtra, India. E-mail: rfernand@chem.iitb.ac.in; Fax: +91-22-25767152; Tel: +91-22-25767174
First published on 11th March 2014
Abstract
An efficient γ-epimerization of γ-vinyl-γ-butyrolactone is studied under palladium catalysis. The building blocks derived there in are used in an efficient stereoselective synthesis of (+)-exo- and (+)-endo-brevicomins.
Introduction
The palladium catalyzed allylic substitution of allyl acetates, allyl carbonates and allyl halides (via the formation of π-allylpalladium complexes) with various nucleophiles are well documented in literature.1 The allylic substitution of unsaturated bridged and fused lactones has been elegantly studied.2 The π-allylpalladium complexes derived from homochiral γ-vinyl-γ-lactones can possibly undergo racemization in the absence of external nucleophiles. The internal carboxylate ion may react with (π-allyl)palladium intermediate and ring-close giving back the lactone. In case of chiral β-substituted γ-vinyl-γ-lactones the recombination can result in syn-anti interconversion products. This mode of ring closure of η3 π-allyl complex has been rarely studied in detail, though an initial pivotal work has been reported by Hon et al.,3 where in epimerization of various substituted cis- and trans-γ-alkenyl-γ-butyrolactones and γ-alkenyl-bisbutyrolactones was observed. In the former case, (±)-1 with β-OTBS group, the epimerization was non selective resulting in approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, syn–anti mixture (Fig. 1). In spite of several advances in allylic substitutions, the Pd-catalyzed syn–anti epimerization for γ-vinyl-γ-lactones through η3 π-allyl species has received little attention. We anticipated a careful interplay of various available Pd-catalysts would result in a better isomer distribution. Considering syn-1a or anti-1b (or 2a and 2b) to have an excellent chemo-differentiated vicinal OH groups: one masked into the lactone and other as protected or free β-OH group, we visualized these as important building blocks in natural product synthesis. The syn-lactone 2a is now available efficiently and reliably by a one-pot three-step sequence from D-glucono-δ-lactone4,5a and has been elegantly used in the protecting-group free synthesis of cardiobutanolide and Hagen's gland lactones.5 The ent-2a is also accessible from L-mannonic-γ-lactone.4 However, the latter is non-natural and expensive (5 g = $ 415.6, Aldrich Chem. Co.). The anti-lactone 2b is not accessible directly. Hence we considered a possibility of direct conversion of syn-1a or 2a to the more stable anti-lactones 1b or 2b respectively using the π-allylpalladium catalysed γ-vinyl-γ-lactone epimerization strategy. While this epimerization is not explored in details the present work describes a detailed optimization in this direction and also further application of the building blocks 1a/1b and 2a/2b in an efficient total synthesis of (+)-exo- and (+)-endo-brevicomins (3 and 4, Fig. 2).6
:1, syn–anti mixture (Fig. 1). In spite of several advances in allylic substitutions, the Pd-catalyzed syn–anti epimerization for γ-vinyl-γ-lactones through η3 π-allyl species has received little attention. We anticipated a careful interplay of various available Pd-catalysts would result in a better isomer distribution. Considering syn-1a or anti-1b (or 2a and 2b) to have an excellent chemo-differentiated vicinal OH groups: one masked into the lactone and other as protected or free β-OH group, we visualized these as important building blocks in natural product synthesis. The syn-lactone 2a is now available efficiently and reliably by a one-pot three-step sequence from D-glucono-δ-lactone4,5a and has been elegantly used in the protecting-group free synthesis of cardiobutanolide and Hagen's gland lactones.5 The ent-2a is also accessible from L-mannonic-γ-lactone.4 However, the latter is non-natural and expensive (5 g = $ 415.6, Aldrich Chem. Co.). The anti-lactone 2b is not accessible directly. Hence we considered a possibility of direct conversion of syn-1a or 2a to the more stable anti-lactones 1b or 2b respectively using the π-allylpalladium catalysed γ-vinyl-γ-lactone epimerization strategy. While this epimerization is not explored in details the present work describes a detailed optimization in this direction and also further application of the building blocks 1a/1b and 2a/2b in an efficient total synthesis of (+)-exo- and (+)-endo-brevicomins (3 and 4, Fig. 2).6
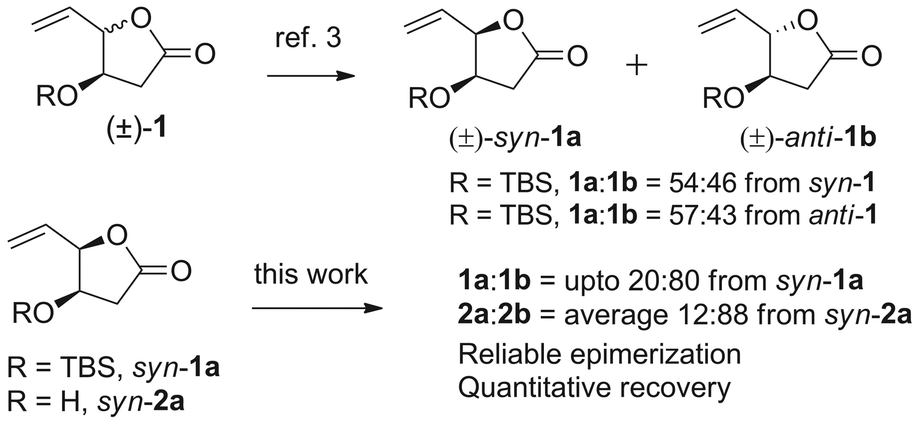 | ||
| Fig. 1 π-Allyl palladium catalysed γ-vinyl-γ-lactone epimerization. | ||
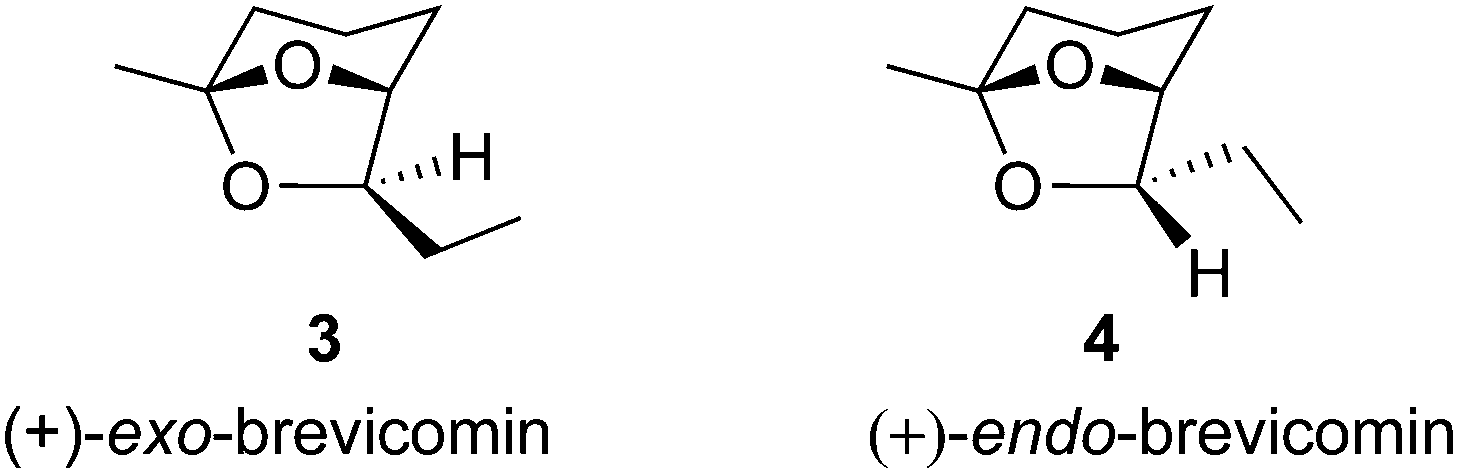 | ||
| Fig. 2 Structures of (+)-exo- and (+)-endo-brevicomins. | ||
Results and discussion
Our investigation actually commenced with the interest to generalize the applications of lactone 1a in various reactions.7 In the Heck reaction of 1a with iodobenzene using catalytic Pd(OAc)2, we observed no Heck coupled product, but an efficient allylic isomerization of 1a to 1b. Though this reaction has a literature precedence,3 it was unexplored hitherto for improved selectivity. Table 1 shows the optimization study of this isomerization using different Pd-catalysts. Diastereomers 1a/1b and 2a/2b were efficiently separable by column chromatography. This enabled to recycle unisomerized 1a or 2a for repeated runs. Initial experiments using different Pd-catalysts (10 mol%, Table 1, entries 1–5) at 50 °C over 72 hours indicated isomerization of pure syn-1a to a mixture, favoring the anti-isomer 1b though marginally from 1:1 up to 1:2. When catalyst loading was increased to 20 mol% (Table 1, entries 6–10), an improved anti-selectivity was observed, with the best result obtained with [PdCl(allyl)]2 giving 1a:1b in 20:80 ratio (Table 1, entry 9) with 80% yield of 1b isolated from the isomerized mixture. We tried reactions for shorter time and at room temperature considering possible early equilibration of the isomers (Table 1, entries 11 and 12). However no further improvement was observed. Next the unprotected β-hydroxy lactone 2a on similar treatment with [PdCl(allyl)]2 (10 mol%) at room temperature over 6 h, gave 2a:2b in 1:2 ratio (Table 1, entry 13). However the use of Pd(OAc)2 (10 mol%, Table 1, entry 14) gave the best isomerization of 2a to 2b in 12:888 ratio with 88% yield of 2b isolated from the isomerized mixture. Lowering of temperature to 10 °C gave inferior results (Table 1, entry 15). Further monitoring the reaction over different time intervals 3, 4, 5, 7 and 8 h (Table 1, entries 16–20) showed that the 6 h reaction (entry 14) was ideal for isomerization. Increase in catalyst loading to 20 mol% and monitoring of time did not yield improved results (Table 1, entries 21–24).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pd catalyst (mol%) | 1a/2a | t (h) | T (°C) | (1a:1b)b or (2a:2b)b |
1b or 2b (%)c |
| a 1a (0.206 mmol), PPh3 (4 equiv. wrt Pd-catalyst), pyridine (0.5 equiv.), THF (3 mL). For 2a (0.4 mmol) and pyridine was not used.b Ratio by 1H NMR.c Isolated yields of 1b or 2b from the isomerized mixture.d Dibenzylidene-acetone impurity was eluted with the product.e PPh3was not used. | ||||||
| 1 | Pd(OAc)2 (10) | 1a | 72 | 50 | 44:56 |
50 |
| 2 | Pd(CF3CO2)2 (10) | 1a | 72 | 50 | 37:63 |
52 |
| 3 | Pd2(dba)3 (10) | 1a | 72 | 50 | 35:65 |
65d |
| 4 | [PdCl(allyl)]2 (10) | 1a | 72 | 50 | 33:67 |
61 |
| 5 | Pd(PPh3)4 (10)e | 1a | 72 | 50 | 38:62 |
53 |
| 6 | Pd(OAc)2 (20) | 1a | 72 | 50 | 29:71 |
71 |
| 7 | Pd(CF3CO2)2 (20) | 1a | 72 | 50 | 49:51 |
28 |
| 8 | Pd2(dba)3 (20) | 1a | 72 | 50 | 42:58 |
49d |
| 9 | [PdCl(allyl)]2 (20) | 1a | 72 | 50 | 20:80 |
80 |
| 10 | Pd(PPh3)4 (20)e | 1a | 72 | 50 | 30:70 |
67 |
| 11 | Pd(OAc)2 (20) | 1a | 6 | rt | 41:59 |
55 |
| 12 | [PdCl(allyl)]2 (20) | 1a | 6 | rt | 40:60 |
57 |
| 13 | [PdCl(allyl)]2 (10) | 2a | 6 | rt | 33:67 |
64 |
| 14 | Pd(OAc)2 (10) | 2a | 6 | rt | 12:888 |
88 |
| 15 | Pd(OAc)2 (10) | 2a | 12 | 10 | 36:64 |
61 |
| 16 | Pd(OAc)2 (10) | 2a | 3 | rt | 34:66 |
65 |
| 17 | Pd(OAc)2 (10) | 2a | 4 | rt | 32:68 |
66 |
| 18 | Pd(OAc)2 (10) | 2a | 5 | rt | 20:80 |
78 |
| 19 | Pd(OAc)2 (10) | 2a | 7 | rt | 15:85 |
82 |
| 20 | Pd(OAc)2 (10) | 2a | 8 | rt | 30:70 |
67 |
| 21 | Pd(OAc)2 (20) | 2a | 3 | rt | 24:76 |
72 |
| 22 | Pd(OAc)2 (20) | 2a | 6 | rt | 25:75 |
74 |
| 23 | Pd(OAc)2 (20) | 2a | 8 | rt | 30:70 |
67 |
| 24 | Pd(OAc)2 (20) | 2a | 32 | rt | 26:74 |
72 |
A plausible mechanism of Pd-catalyzed epimerization is rationalized in Scheme 1. The opening of lactone 2a give the π-allyl intermediate A. Further single bond rotation will lead to B which gets stabilization through OH–Pd(0) interaction as in C.9 Subsequent ring closure from opposite face of Pd-coordination will result in the epimerized lactone 2b. While β-OTBDMS containing lactone requires higher catalyst loading (20 mol%, entry 9, Table 1) due to steric crowding, the free β-OH containing lactone epimerized with 10 mol% catalyst loading (entry 14) due to better OH–Pd(0) interaction.
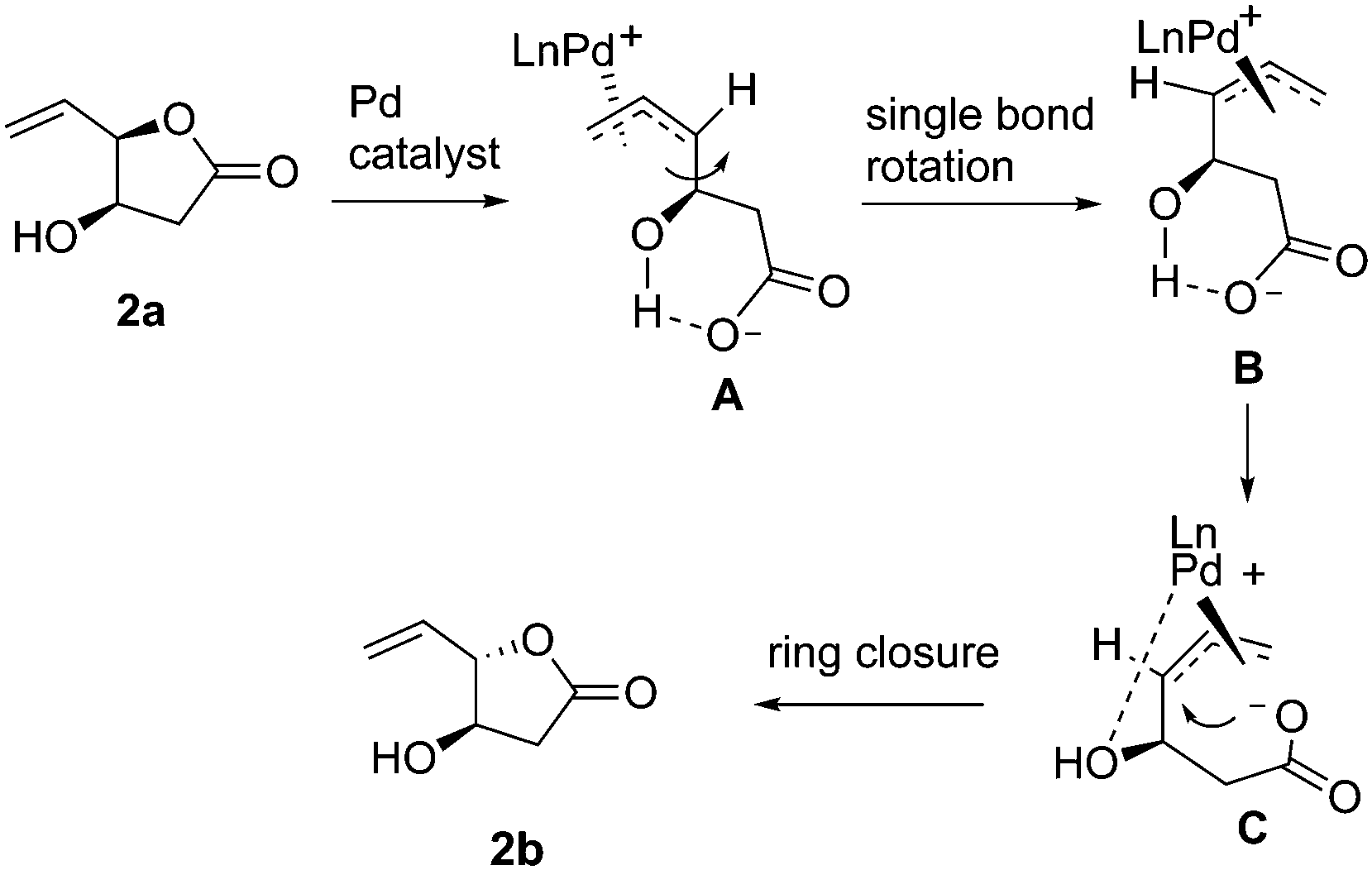 | ||
| Scheme 1 Plausible mechanism of the Pd-catalyzed epimerization of lactone 2a. | ||
With the synthesized and epimerized building block lactones 1a, 1b, 2a and 2b in hand, we turned our focus on using them in the synthesis of (+)-exo- and (+)-endo-brevicomins (3 and 4, Fig 2). The brevicomins 3 and 4 are volatile components of the attracting pheromones of several bark beetle species of the Dendroctonus and Dryocetes family.10 Exo-brevicomin is an aggregation pheromone of the western pine beetle, Dendroctonus brevicomis and endo-brevicomin is a minor component accompanying exo-brevicomin and is antiaggregation pheromone of the southern pine beetles.11 D. brevicomis is a serious pest causing destruction in the timber regions of the west coast of North America. Exo-brevicomin can be used in the population control of this destructive insect by manipulating its mating habits.12 The synthesis of (+)-exo- and (+)-endo-brevicomins (3 and 4) is shown in Scheme 2. D-Glucono-δ-lactone (5) was processed through a one-pot procedure into β-hydroxy-γ-vinyl-γ-butyrolactone 2a in 51% yield.5 Silyl protection of free hydroxyl group to 1a (86%) and subsequent DIBAL-H reduction afforded the lactol which was immediately treated with Wittig ylide, Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOCH3 in toluene solvent at reflux to give the unstable compound 6. Attempts to isolate pure 6 were unsuccessful as it undergoes a facile conjugate addition of free hydroxyl group to the α,β-unsaturated ketone unit giving a mixture of compounds. Hence the crude 6 after removal of toluene solvent was hydrogenated in EtOH using 10% Pd/C to afford 7 in overall 37% yield. This reaction was further optimized with the Wittig reaction in dichloromethane solvent under reflux condition for 30 h followed by hydrogenation of the crude product 6 in iPrOH/EtOAc (3:8) using Pd(OH)2/C, resulting in an improved yield of 7 (68% from 1a). The anti-lactone 2b obtained quantitatively after isomerization of 2a (Table 1, entry 14) was silyl protected to give 1b (88%). This was then transformed into keto compound 9 in 71% overall yield from 1b. Removal of silyl group in 7 by HCl hydrolysis in MeOH resulted in spontaneous stereoselective ketal formation delivering (+)-exo-brevicomin 3 in 88% yield, [α]25D = +72.9 (c = 1.2, Et2O); lit.13a [α]20D = +76.3 (c = 1.35, Et2O). Similarly, the keto compound 9 delivered, (+)-endo-brevicomin 4 in 86% yield, [α]20D = +73.3 (c = 1.2, Et2O); lit.13a [α]20D = +77.9 (c = 1.2, Et2O). The spectral and analytical data of 3 and 4 were in excellent agreement with reported data.13
CHCOCH3 in toluene solvent at reflux to give the unstable compound 6. Attempts to isolate pure 6 were unsuccessful as it undergoes a facile conjugate addition of free hydroxyl group to the α,β-unsaturated ketone unit giving a mixture of compounds. Hence the crude 6 after removal of toluene solvent was hydrogenated in EtOH using 10% Pd/C to afford 7 in overall 37% yield. This reaction was further optimized with the Wittig reaction in dichloromethane solvent under reflux condition for 30 h followed by hydrogenation of the crude product 6 in iPrOH/EtOAc (3:8) using Pd(OH)2/C, resulting in an improved yield of 7 (68% from 1a). The anti-lactone 2b obtained quantitatively after isomerization of 2a (Table 1, entry 14) was silyl protected to give 1b (88%). This was then transformed into keto compound 9 in 71% overall yield from 1b. Removal of silyl group in 7 by HCl hydrolysis in MeOH resulted in spontaneous stereoselective ketal formation delivering (+)-exo-brevicomin 3 in 88% yield, [α]25D = +72.9 (c = 1.2, Et2O); lit.13a [α]20D = +76.3 (c = 1.35, Et2O). Similarly, the keto compound 9 delivered, (+)-endo-brevicomin 4 in 86% yield, [α]20D = +73.3 (c = 1.2, Et2O); lit.13a [α]20D = +77.9 (c = 1.2, Et2O). The spectral and analytical data of 3 and 4 were in excellent agreement with reported data.13
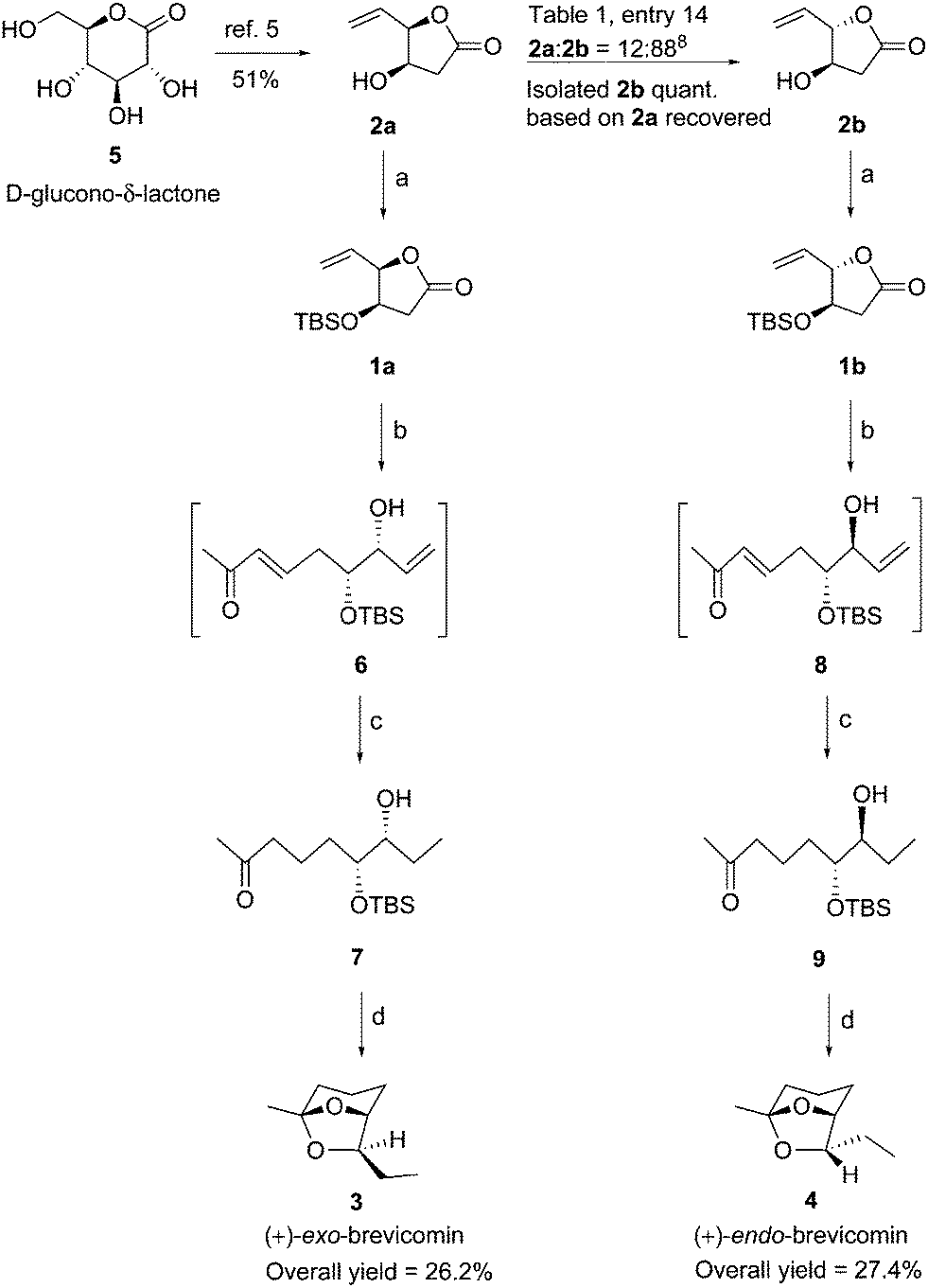 | ||
| Scheme 2 Synthesis of (+)-exo-brevicomin and (+)-endo-brevicomin. Reagents and conditions: (a) TBDMSCl (1.5 equiv.), imidazole (2.0 equiv.), CH2Cl2, 0 °C to room temp., 12 h, 1a (86%), 1b (88%); (b) (i) DIBAL-H (1.5 equiv.), CH2Cl2, −78 °C, 45 min; (ii) Ph3PCH2COCH3 (2.0 equiv.), CH2Cl2, reflux, 30 h; (c) H2, Pd(OH)2/C, iPrOH/EtOAc, room temp., 24 h, 7 (68% from 1a), 9 (71% from 1b); (d) 3N HCl, MeOH, room temp., 2.5 h, 3 (88%), 4 (86%). | ||
Conclusions
In summary, we have explored an efficient Pd-catalyzed γ-epimerization of γ-vinyl-γ-butyrolactone to excess different chiral building blocks. These were then used in the stereoselective synthesis of (+)-exo- and (+)-endo-brevicomins. Thus this remarks an efficient stereodivergent synthesis of the pheromones. The (+)-exo- and (+)-endo-brevicomins were obtained efficiently in 26.2 and 27.4% overall yields, respectively. The strategy explored can be extended to γ-alkenylated γ-lactones or higher lactones to give further unexplored building blocks for natural products synthesis.Experimental section
General information
Flasks were oven or flame dried and cooled in a desiccator. Dry reactions were carried out under an atmosphere of Ar or N2. Solvents and reagents were purified by standard methods. Thin-layer chromatography was performed on EM 250 Kieselgel 60 F254 silica gel plates. The spots were visualized by staining with KMnO4 or under UV lamp. 1H and 13C NMR were recorded with a Bruker, AVANCE III 400 spectrometer and the chemical shifts are based on TMS peak at δ = 0.00 pm for proton NMR and CDCl3 peak at δ = 77.00 ppm (t) in carbon NMR. IR spectra were obtained on Perkin Elmer Spectrum One FT-IR spectrometer and samples were prepared by evaporation from CHCl3 on CsBr plates. Optical rotations were measured with Jasco P–2000 digital polarimeter. High-resolution mass spectra (HRMS) were obtained using positive electrospray ionization by TOF method.(4R,5R)-4-Hydroxy-5-vinyldihydrofuran-2(3H)-one (2a)5a
The titled compound was prepared by the reported literature procedure.5a Data for 2a: [α]25D = +45.3 (c = 0.8, CHCl3). IR (CHCl3): νmax = 3447, 2934, 1771, 1639, 1432, 1413, 1333, 1309, 1203, 1158, 1080, 1017, 990, 962, 901, 884, 833, 796 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 5.96 (ddd, J = 17.4, 10.7, 6.0 Hz, 1H), 5.57 (dt, J = 17.4, 1.3 Hz, 1H), 5.52 (dt, J = 10.7, 1.2 Hz, 1H), 4.94–4.91 (m, 1H), 4.56–4.53 (m, 1H), 2.80 (dd, J = 17.7, 5.4 Hz, 1H), 2.63 (dd, J = 17.7, 1.3 Hz, 1H), 2.27 (brs, 1H, OH) ppm. 13C NMR (100 MHz, CDCl3) δ = 176.1, 130.2, 120.6, 84.9, 69.4, 38.6 ppm. HRMS m/z calcd for [C6H8O3 + H]+ 129.0552, found 129.0551.(4R,5R)-4-(tert-Butyldimethylsilyloxy)-5-vinyldihydrofuran-2(3H)-one (1a)
To a solution of hydroxy lactone 2a (0.5 g, 3.902 mmol) in dry CH2Cl2 (15 mL) was added imidazole (0.531 g, 7.804 mmol, 2.0 equiv.), followed by tert-butyldimethylsilyl chloride (0.882 g, 5.853 mmol, 1.5 equiv.) at 0 °C. The reaction mixture was stirred at room temperature for 12 h and then diluted with CH2Cl2 (10 mL) and H2O (10 mL). The solution was extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were washed with water, brine, dried (Na2SO4) and concentrated. The residue was purified by silica gel column chromatography using petroleum ether–EtOAc (9:1) as eluent to afford 1a (0.813 g, 86%) as a colorless oil. [α]25D = +15.9 (c = 1.0, CHCl3). IR (CHCl3): νmax = 2956, 2932, 2859, 1781, 1473, 1259, 1206, 1149, 1096, 1029, 946, 841, 779 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 5.97 (ddd, J = 17.4, 10.4, 7.2 Hz, 1H), 5.42 (dt, J = 17.4, 1.1 Hz, 1H), 5.37 (dt, J = 10.5, 1.0 Hz, 1H), 4.79 (dd, J = 7.2, 4.2 Hz, 1H), 4.51–4.48 (m, 1H), 2.73 (dd, J = 17.2, 5.4 Hz, 1H), 2.48 (dd, J = 17.2, 2.1 Hz, 1H), 0.87 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ = 175.2, 131.8, 119.9, 85.4, 70.7, 39.3, 25.6, 18.0, −4.9, −5.0 ppm. HRMS m/z calcd for [C12H22O3Si + H]+ 243.1415, found 243.1418.
(4R,5S)-4-(tert-Butyldimethylsilyloxy)-5-vinyldihydrofuran-2(3H)-one (1b)
:1b = 20:80 mixture (by 1H NMR). The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (19:1) as eluent to afford 1b (40 mg, 80% yield, quantitative based on recovered 1a) as a colorless oil and further elution gave the starting material 1a (10 mg, 20%). Data for 1b: [α]25D = −34.7 (c = 0.6, CHCl3). IR (CHCl3): νmax = 2956, 2931, 2889, 2859, 1791, 1473, 1408, 1362, 1261, 1201, 1163, 1084, 1034, 941, 909, 891, 839, 780 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 5.82 (ddd, J = 17.0, 10.8, 6.0 Hz, 1H), 5.39 (d, J = 17.2 Hz, 1H), 5.29 (d, J = 10.6 Hz, 1H), 4.72–4.69 (m, 1H), 4.26–4.22 (m, 1H), 2.73 (dd, J = 17.4, 6.4 Hz, 1H), 2.42 (dd, J = 17.4, 4.3 Hz, 1H), 0.88 (s, 9H), 0.075 (s, 3H), 0.070 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ = 174.7, 132.9, 118.2, 87.6, 72.6, 37.4, 25.6, 17.9, −4.84, −4.86 ppm. HRMS m/z calcd for [C12H22O3Si + Na]+ 265.1236, found 265.1241.(4R,5S)-4-Hydroxy-5-vinyldihydrofuran-2(3H)-one (2b)
:2b = 12:88 mixture (by 1H NMR).8 The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (7:3) as eluent to afford 2b (45.1 mg, 88%, quantitative based on recovered 2a) as a colorless oil. Further elution gave the starting material 2a (6.2 mg, 12%) as colorless oil. Data for 2b: [α]25D = −25.0 (c = 1.0, CHCl3). IR (CHCl3): νmax = 3445, 2932, 2852, 1771, 1646, 1416, 1360, 1265, 1179, 1101, 1061, 990, 936, 879 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 5.85 (ddd, J = 17.1, 10.7, 5.3 Hz, 1H), 5.41 (d, J = 17.2 Hz, 1H), 5.29 (d, J = 10.7 Hz, 1H), 4.86–4.84 (m, 1H), 4.38–4.35 (m, 1H), 2.79 (dd, J = 17.8, 6.2 Hz, 1H), 2.55 (brs, 1H, OH), 2.50 (dd, J = 17.8, 3.3 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ = 175.0, 132.7, 118.3, 87.2, 71.8, 36.6 ppm. HRMS m/z calcd for [C6H8O3 + H]+ 129.0552, found 129.0555.(4R,5S)-4-(tert-Butyldimethylsilyloxy)-5-vinyldihydrofuran-2(3H)-one (1b)
The titled compound was prepared from 2b (0.100 g, 0.780 mmol) by similar procedure as described for 1a, to give 1b (0.166 g, 88%) as colorless oil. [α]25D = −36.2 (c = 0.6, CHCl3). Other analytical data were same as before.(6R,7R)-6-(tert-Butyldimethylsilyloxy)-7-hydroxynonan-2-one (7)
To a stirred solution of lactone 1a (0.2 g, 0.825 mmol) in dry CH2Cl2 (15 mL) was added DIBAL-H (1.75 M solution in toluene, 0.73 mL, 1.24 mmol, 1.5 equiv.) drop wise over a period of 10 min at −78 °C. The reaction mixture was stirred for 45 min and then quenched with a saturated aq. solution of Rochelle's salt (5 mL) at −78 °C. The stirring was continued for 2 h at room temperature and the solution extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with water, brine, dried (Na2SO4) and concentrated to give the crude lactol (0.2 g) which was used for the next reaction without further purification.To the solution of above lactol (0.2 g) in dry CH2Cl2 (15 mL) was added 1-(triphenylphosphoranylidene)-2-propanone (Ph3PCHCOCH3) (0.525 g, 1.65 mmol, 2.0 equiv.) and the solution heated to reflux for 30 h. The reaction mixture was cooled to room temperature and the solvent was removed under reduced pressure. The resulting residue was dissolved in iPrOH/AcOEt (5 mL, 3:8) and treated with 10% Pd(OH)2/C (22 mg). The reaction mixture was vigorously stirred under H2 (balloon pressure) atmosphere at room temperature for 24 h. The mixture was filtered through a pad of Celite and silica gel, washed with EtOAc and concentrated. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (4:1) as eluent to give 7 (0.162 g, 68% over three steps) as a colorless oil. [α]25D = −11.1 (c = 1.6, CHCl3). IR (CHCl3): νmax = 3460, 2957, 2932, 2885, 2859, 1716, 1472, 1464, 1361, 1256, 1163, 1094, 1071, 837, 776, 674 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 3.54–3.52 (m, 1H), 3.38–3.32 (m, 1H), 2.45–2.41 (m, 2H), 2.13 (s, 3H), 2.08 (d, J = 7.4 Hz, 1H), 1.62–1.54 (m, 4H), 1.47–1.40 (m, 2H), 0.97 (t, J = 7.4 Hz, 3H), 0.90 (s, 9H), 0.08 (s, 3H), 0.07 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ = 208.6, 74.4, 74.0, 43.7, 33.3, 29.8, 26.9, 25.8, 19.3, 18.0, 10.4, −4.2, −4.7 ppm. HRMS m/z calcd for [C15H32O3Si + Na]+ 311.2018, found 311.2023.
(6R,7S)-6-(tert-Butyldimethylsilyloxy)-7-hydroxynonan-2-one (9)
The title compound was prepared from 1b (0.2 g, 0.825 mmol) by a similar procedure as described for 7, to give 9 (0.169 g, 71% over three steps) as colorless oil. [α]25D = +5.6 (c = 0.3, CHCl3). IR (CHCl3): νmax = 3480, 2957, 2931, 2885, 2858, 1716, 1464, 1407, 1361, 1255, 1163, 1094, 1048, 977, 837, 776 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 3.63–3.59 (m, 1H), 3.52–3.48 (m, 1H), 2.45–2.41 (m, 2H), 2.19 (d, J = 2.9 Hz, 1H), 2.13 (s, 3H), 1.73–1.37 (m, 6H), 0.97 (t, J = 7.4 Hz, 3H), 0.90 (s, 9H), 0.077 (s, 3H), 0.074 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ = 208.9, 75.9, 74.8, 43.8, 30.0, 29.9, 25.8, 24.8, 19.9, 18.0, 10.6, −4.5 ppm. HRMS m/z calcd for [C15H32O3Si + Na]+ 311.2018, found: 311.2022.(+)–exo–Brevicomin (3)
To a stirred solution of 7 (120 mg, 0.416 mmol) in methanol (8 mL) was added 3 N HCl (0.5 mL) at room temperature and it was stirred for 2.5 h. The reaction mixture was diluted with Et2O (10 mL) and H2O (5 mL) and the aqueous phase was separated and extracted with Et2O (2 × 5 mL). The combined organic layers were washed with water, brine, dried (Na2SO4) and concentrated. The residue was purified by silica gel column chromatography using petroleum ether/Et2O (49:1) as eluent to give 3 (57 mg, 88%) as a colorless oil. [α]25D = +72.9 (c = 1.2, Et2O); lit.13a [α]20D = +76.3 (c = 1.35, Et2O). IR (CHCl3): νmax = 2938, 2868, 1737, 1652, 1514, 1463, 1383, 1350, 1331, 1238, 1195, 1174, 1107, 1029, 1006, 969, 879, 846 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 4.12 (brs, 1H), 3.92 (t, J = 6.5 Hz, 1H), 1.91–1.73 (m, 2H), 1.62–1.45 (m, 6H), 1.40 (s, 3H), 0.89 (t, J = 7.5 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ = 107.7, 81.1, 78.2, 34.9, 28.5, 27.9, 25.0, 17.1, 9.7 ppm. HRMS m/z calcd for [C9H16O2 + H]+ 157.1229, found 157.1224.
(+)–endo–Brevicomin (4)
The title compound was prepared from 9 (100 mg, 0.347 mmol) by a similar procedure as described for 3, to give 4 (46.5 mg, 86%) as a colorless oil. [α]25D = +73.3 (c = 1.2, Et2O); lit.13a [α]20D = +77.9 (c = 1.2, Et2O). IR (CHCl3): νmax = 2960, 2936, 2878, 1728, 1514, 1465, 1379, 1349, 1309, 1260, 1238, 1173, 1108, 1032, 1001, 967, 902, 870, 852 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 4.21–4.20 (m, 1H), 4.00–3.96 (m, 1H), 1.95–1.77 (m, 2H), 1.69–1.53 (m, 6H), 1.43 (s, 3H), 0.98 (t, J = 7.5 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ = 107.0, 81.6, 76.5, 34.4, 25.0, 23.6, 21.9, 17.5, 11.0 ppm. HRMS m/z calcd for [C9H16O2 + H]+ 157.1229, found 157.1226.Acknowledgements
We thank the Department of Science and Technology, New Delhi (grant no. SR/S1/OC-25/2008) and Department of Chemistry, IIT-Bombay for financial support. P. K. and V. B. thank the Council of Scientific and Industrial Research (CSIR) New Delhi for research fellowships.Notes and references
- For selected reviews for Pd-catalyzed allylic substitutions, see: (a) B. M. Trost and D. L. van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS PubMed; (c) J. Tsuji, in Palladium Reagents and Catalysts: New Perspectives for the 21st Century, John Wiley and Sons, Ltd., Chichester, U.K., 2004, ch. 4, p. 431 Search PubMed; (d) B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467–4483 RSC.
- V. K. Aggarwal, N. Monteiro, G. J. Tarver and R. McCague, J. Org. Chem., 1997, 62, 4665–4671 CrossRef CAS and references cited therein.
- Y.-S. Hon, H.-F. Chen, C.-Y. Kao and C.-Z. Luo, Tetrahedron, 2010, 66, 8468–8475 CrossRef CAS PubMed.
- J. Song and R. I. Hollingsworth, Tetrahedron: Asymmetry, 2001, 12, 387–391 CrossRef CAS.
- (a) R. A. Fernandes and P. Kattanguru, Asian J. Org. Chem., 2013, 2, 74–84 CrossRef CAS PubMed ; A provisional process patent on this work has been filed, application no. 1780/MUM/2012; (b) R. A. Fernandes and P. Kattanguru, J. Org. Chem., 2012, 77, 9357–9360 CrossRef CAS PubMed ; A provisional process patent on this work is filed, application no. 1908/MUM/2012.
- K. Mori, Biosci., Biotechnol., Biochem., 2011, 75, 976–981 CrossRef CAS and references cited therein.
- R. A. Fernandes and P. Kattanguru, unpublished results.
- The ratio of 2a:2b ranged from 9:91 to 14:86. The average of 6 reactions was found to be 12:88.
- K. Hallman, A. Frölander, T. Wondimagegn, M. Svensson and C. Moberg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5400–5404 CrossRef CAS PubMed.
- (a) R. M. Silverstein, R. G. Brownlee, T. E. Bellas, D. L. Wood and L. E. Browne, Science, 1968, 159, 889–891 CAS; (b) W. D. Bedard, P. E. Tilden, D. L. Wood, R. M. Silverstein, R. G. Brownlee and J. O. Rodin, Science, 1969, 164, 1284–1285 CAS; (c) D. L. Wood, L. E. Browne, B. Ewing, K. Lindahl, W. D. Bedard, P. E. Tilden, K. Mori, G. B. Pitman and P. R. Hughes, Science, 1976, 192, 896–898 CAS.
- J. P. Vité, R. F. Billings, C. W. Ware and K. Moro, Naturwissenschaften, 1985, 72, 99–100 CrossRef.
- W. D. Bedard and D. L. Wood, Programs utilizing pheromones in survey and control. Bark beetles-the western pine beetle, in Pheromones, ed. M. C. Birch, North-Holland, Amsterdam, 1974, pp. 441−449 Search PubMed.
- (a) S. Singh and P. J. Guiry, J. Org. Chem., 2009, 74, 5758–5761 CrossRef CAS PubMed; (b) P. Pal and A. K. Shaw, Tetrahedron, 2011, 67, 4036–4047 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all the compounds. See DOI: 10.1039/c4ra00814f |
| This journal is © The Royal Society of Chemistry 2014 |