 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of the alkali metal cation on the electrocatalytic oxidation of formate on platinum
Bruno A. F. Previdelloa,
Eduardo G. Machadoa and
Hamilton Varela*ab
aInstitute of Chemistry of São Carlos, University of São Paulo, PO Box 780, São Carlos, 13560-970, SP, Brazil. E-mail: varela@iqsc.usp.br
bFritz Haber Institute of the Max Planck Society, Department of Physical Chemistry, Faradayweg 4-6, D-14195 Berlin, Germany
First published on 14th March 2014
Abstract
Non-covalent interactions between hydrated alkali metal cations and adsorbed oxygenated species on platinum might considerably inhibit some electrocatalytic reactions. We report in this communication the effect exerted by electrolyte alkali metal cations on the electro-oxidation of formate ions on platinum. The system was investigated by means of cyclic voltammetry and chronoamperometry in the presence of an electrolyte containing Li+, Na+, or K+. As already observed for other systems, the general activity towards the electro-oxidation of formate ions was found to increase in the sequence Li+ < Na+ < K+. In addition, we observed that the inhibition caused by smaller cations has a peculiar potential dependence because of the multi-peaked current profile of the electro-oxidation of formate on platinum. In this respect, we have also identified a new effect caused by cation inhibition at intermediate potentials, namely a peak splitting towards the use of smaller cations. Results are discussed in connection with mechanistic aspects of this model system.
1. Introduction
It has been recently shown that the nature of the alkali metal cation in the electrolyte severely impacts the electro-oxidation of molecules such as carbon monoxide,1 ethylene glycol,2,3 glycerol,3 hydrogen,4 methanol,4 and the reduction of oxygen.5 Additionally, the potential range of the transition region between diffusion-limited reduction to oxidation of hydrogen peroxide is also dependent on the alkali cation, as stated by Mayrhofer et al.6The importance of the so-called non-covalent interactions between hydrated alkali metal cations and adsorbed hydroxyl species on the platinum surface was brought to light by Marković and co-workers.4 The presence of OHad–M+(H2O)x clusters at the interface increases in the order of the hydration energy of the alkali metal M (i.e., Li+ > Na+ > K+ > Cs+) and a consequent surface blockage is observed in the same order.4 For most systems, the electrocatalytic activity increases in the sequence: Li+ < Na+ < K+ < Cs+.
The electro-oxidation of formate ions on platinum has been studied in different contexts.7–12 Osawa and co-workers8 investigated the electro-oxidation of formic acid/formate in the pH range between 0 and 12 and established that formate ion is the key reactant over the whole pH window. They argued on the importance of the acid–base equilibrium on the process and observed that the closer the solution pH is of the pKa of formic acid, the greater is the oxidation current. Abruña and co-workers10 have recently discussed the mechanism of formate electro-oxidation thoroughly. The authors stated that the process starts with the adsorption of a given X1 species, which is a stable adsorbate that adsorbs more weakly than formate itself. This adsorbate may react by the primary pathway, which leads directly to CO2 or by the secondary pathway, in which X1 generates COads that reacts with adsorbed OH− to CO2. Wieckowski and co-workers7 discussed the occurrence of three peaks along the potentiodynamic sweep as resulting of a triple pathway mechanism, including a third path through the oxidation of less-active adsorbate at higher potentials in addition to the well-known dual pathway. From the applied perspective, the electro-oxidation of formate ions is of utter importance, because of the increasingly interest in alkaline fuel cells.13–15
Motivated by the intricate multi-processes feature of the current–potential curve of the electro-oxidation of formate ions on platinum, herein we present results on the experimental investigation of the effect of the nature of the alkali metal cation in the electrolyte on this system.
2. Experiments and methods
Solutions
All solutions were prepared with ultra-pure Millipore® water (18.2 MΩ cm). Aqueous solutions at 0.1 M of KOH (Sigma Aldrich), NaOH (Sigma-Aldrich), or LiOH (Sigma-Aldrich) were used as electrolytes. Prior to each experiment, formic acid (SigmaAldrich) was added to the electrolyte solution in order to yield formate at the concentration of 0.1 M.Electrodes
The working electrode was a polycrystalline platinum flag with geometric area of 0.302 cm2. All current densities are referred to the electroactive area, as measured before each experiment by means of the hydrogen oxidation. The electrode roughness was typically about 1.42. The counter electrode was a platinum net with very high surface area. The reference electrode was a reversible hydrogen electrode prepared with the supporting electrolyte, with respect to which all potentials will be referred to.Cleanness
Since working with alkaline media demands a special attention to the cleanness of the system as to ensure the reproducibility of the results, before each experiment, a chemical cleaning was employed. All the glassware was immersed in a slightly alkaline potassium permanganate solution the day before the experiment. In the next day, it was washed with water and immersed in slightly acid hydrogen peroxide solution. Next, it was washed thoroughly with boiling ultra-pure water. After the chemical cleaning, the electrode was flame-annealed with a butane flame for about 1 minute and then cooled at argon atmosphere. Finally, it was inserted into the cell and it was performed an electrochemical annealing, with 150 voltammetric cycles ranging from 0.05 V to 1.5 V at 2 V s−1.Electrochemical setup
The cell used was a one-chambered cell and all experiments were performed with stagnant solution. Before and during all measurements, argon (White Martins®) was purged into the system in order to reduce oxygen dissolution. Experiments were performed at 25 ± 1 °C, controlled by a thermostat. The electrochemical measurements were accomplished using an Autolab® potentiostat (model PGSTAT302N) with the modulus Scan-Gen coupled (analog scan generator).Deconvolution
The deconvolution of the voltammetric peaks was performed with the software Origin 8.6®. Initially it was drawn a baseline for the first half of the scan, in order to eliminate experimental noise and minimize the capacitance component of the total current. Next, the peaks were selected and it was performed a Gaussian curve fitting. The coefficient of determination (R2) for the fitting ranged from 0.990 to 0.996.3. Results and discussion
Cyclic voltammetry
Fig. 1 displays the (a) base-cyclic voltammograms, (b) the positive-, and (c) the negative-going sweep after addition of formate ions. The base-CVs are identical to that previously reported2 and the main aspect to be highlighted is the fact that the non-covalent interactions slightly postpones the surface oxidation as clearly seen along the positive and, more transparently, the negative-going sweep, where the reduction charge increase in the sequence Li+ < Na+ ∼ K+. As already mentioned, this observation results from the higher interfacial concentration of OHad–M+(H2O)x clusters accompanying the increase in the hydration energy or the decrease in the ionic radius of the alkali metal cations.4 After this initial inhibition on the onset of surface oxidation, the voltammetric profiles become very similar, as the interactions between hydrated cation and the platinum surface are negligible at E > ∼1.0 V. This is due to the cation repulsion at high potentials and also to the prevailing of the interaction between platinum and oxygenated species.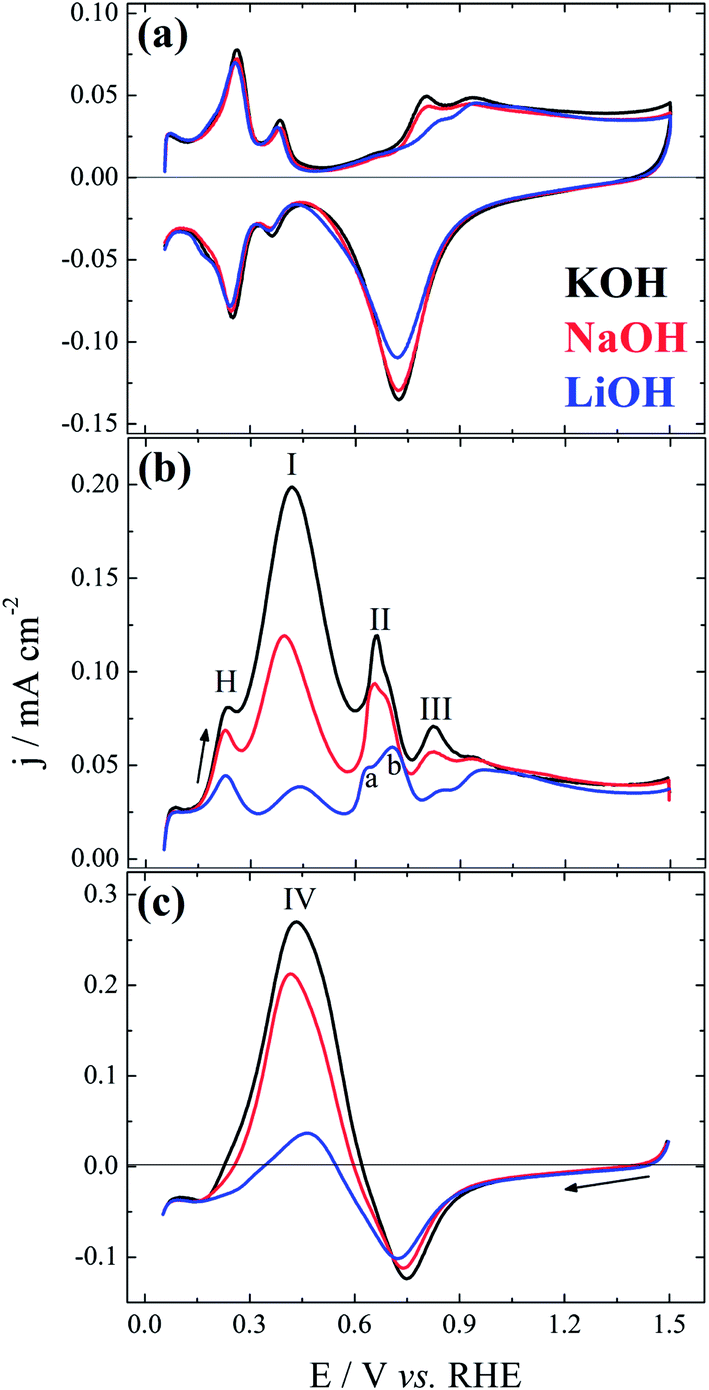 | ||
| Fig. 1 Voltammetric characterization of the platinum electrode in (a) 0.1 MOH (M = K, Na, Li); and after addition of 0.1 M HCOO− along the (b) positive-, and (c) negative-going sweep. dE/dt = 50 mV s−1. | ||
Focusing now on the voltammetric profiles of the electro-oxidation of formate ions, Fig. 1(b) and (c), the first aspect to be highlighted is the clear activity increase in the sequence Li+ < Na+ < K+. Nevertheless, unlike the similar effect observed for methanol,4 ethylene glycol,2 and glycerol,3 the highly structured or multi-peaked curve observed here makes the effect more intricate. Peak H in Fig. 1(b) can be mainly assigned to hydrogen desorption and it is considerably inhibited by the presence of Li+, and also of Na+ to a lesser extent. Following the notation used by Wieckowski and co-workers,7 peaks I, II, and III are assigned in Fig. 1(b). As further discussed below, peak II is clearly divided into a and b as a smaller cation is used. Peak I has been commonly accepted as relative to the direct oxidation of formate to CO2 without forming COad as intermediate.7,10 The activity decrease observed here can be simply related to the decrease in the surface sites for the adsorption of the initial active intermediate, due to the presence of OHad–M+(H2O)x clusters. Peak II have been assigned to the electro-oxidation of adsorbed carbon monoxide in the indirect pathway.7,10 In that pathway, an intermediate of formate adsorbs and yields COad that reacts with OHad to CO2.
In the modified triple-path scheme, Wieckowski and collaborators7 suggest a third pathway around peak III, completely independent of the two other pathway, involving a less active adsorbate. The fact that peak III is apparently suppressed in the presence of Li+, as suggested by the coincidence in the initial surface oxidation in Fig. 1(a) and (b), is apparently similar to the facilitated COad oxidation as presented by Koper and co-workers.1 The promoting effect is caused by the early adsorption of OH species on the platinum, which favors the oxidation of COad. In the case of Li+, the authors observed that oxidation does not start earlier but it is rather completed at a lower potential, as compared to the situation with Na+ and K+.1 Wieckowski et al.7 suggested that the electro-oxidation of this less active adsorbate would also proceed via a Langmuir-Hishelwood step with an adsorbed oxygenated species. Therefore, a similar effect could be operative here. Further experiments, in particular with well-defined surfaces under comparable conditions, would be needed to clarify this point.
Fig. 1(c) shows the negative-going sweep that follows the forward scan presented in part (b). Peak IV can be primarily related to process observed in peak I,7 but it is also influenced by the autocatalytic production of free platinum sites along oxide reduction.16 The effect of the alkali metal cation on the oxide formation along the positive going sweep is clearly seen in the reduction process characterized by the cathodic peak around 0.75 V. Indeed, the reduction charge increases in the sequence Li+ < Na+ < K+, in the same way than seen in base electrolyte, Fig. 1(a). Once the platinum oxides are reduced, the cation inhibition starts playing a role again, as clearly seen in the increase of peak IV, and also in the potential at which the current becomes positive along the negative-going sweep.
Fig. 2 shows the deconvolution of the current structures presented along the positive-going sweep in Fig. 1(b). The activity trend K+ > Na+ > Li+ is followed for peaks I, IIa, and III. The situation changes for peak IIb: the relative size between peaks IIa and IIb decrease in the sequence K+ > Na+ > Li+, and it becomes smaller than 1 for LiOH electrolyte. Interestingly, the increase in peak IIb seems correlated with the decrease in peak III, in line with the anticipation of the oxidation process in peak III or its earlier completion as caused by smaller cations.
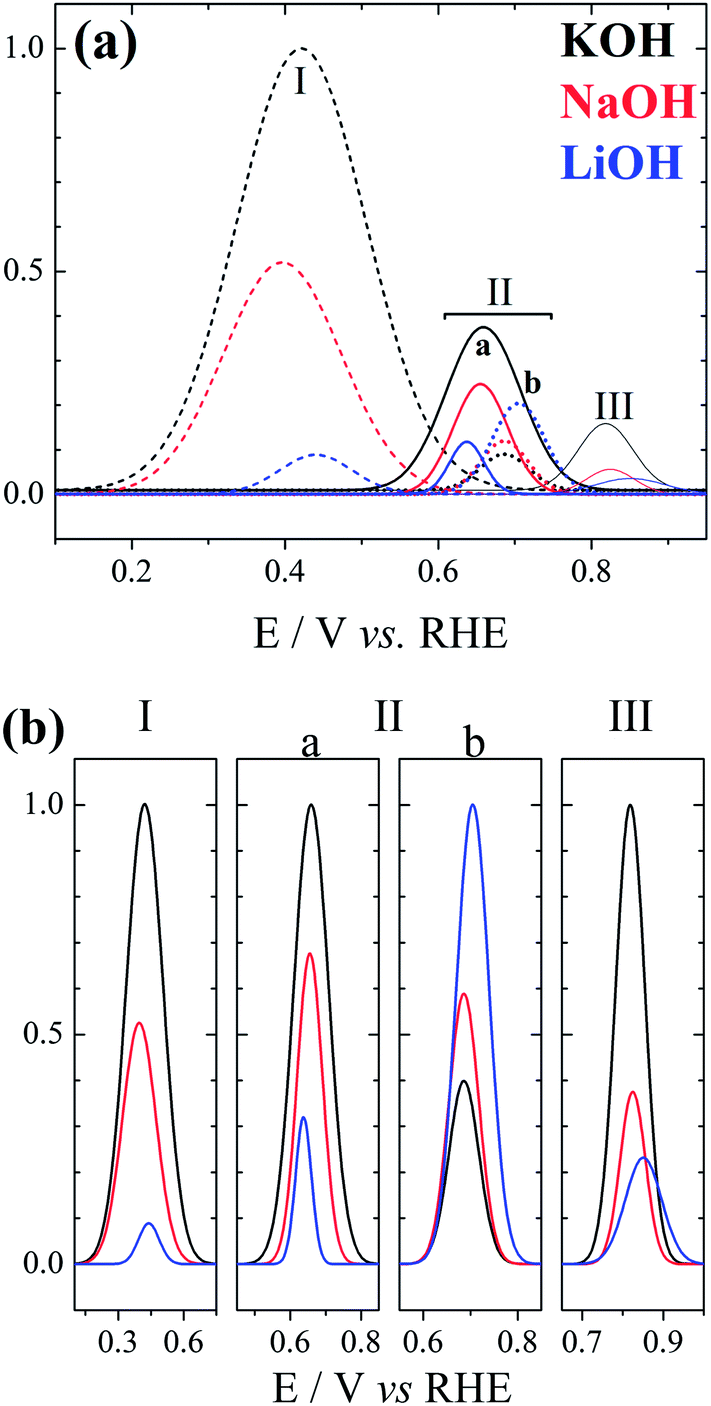 | ||
| Fig. 2 (a) Deconvolution normalized by the higher peak (KOH peak I) of the positive-going sweep profiles presented in Fig. 1(b). (b) Comparison of normalized deconvoluted peaks. | ||
Chronoamperometry
Additionally to the voltammetric investigation just presented, the system was studied by chronoamperometry in order to clarify the role of the noncovalent interaction of alkali metal cations under quasi-stationary regime. Current–time evolutions were monitored at different potentials after a potential step from a certain initial value. This initial potential, E0, was either 0.05 V or 1.20 V. Fig. 3(a) shows typical current profiles at E = 0.40 V for three electrolytes and for two initial potentials. Fig. 3(b) summarizes the total oxidation charge, i.e. 0 < t < 300 s, for all experiments. The activity trend follows what have been observed in the cyclic voltammograms in Fig. 1(b) and (c). The first aspect to be discussed here is that, in almost all cases, the overall activity is higher when the electrode is initially polarized at 1.20 V. In fact, at such high potential the surface is nearly free of carbonaceous poison17 but also from OHad–M+(H2O)x clusters. As a consequence, the inhibition caused by smaller hydrated cations is less pronounced when the initial potential is 1.20 V. For E0 = 0.05 V, the surface started already poisoned by carbonaceous fragments and OHad–M+(H2O)x clusters.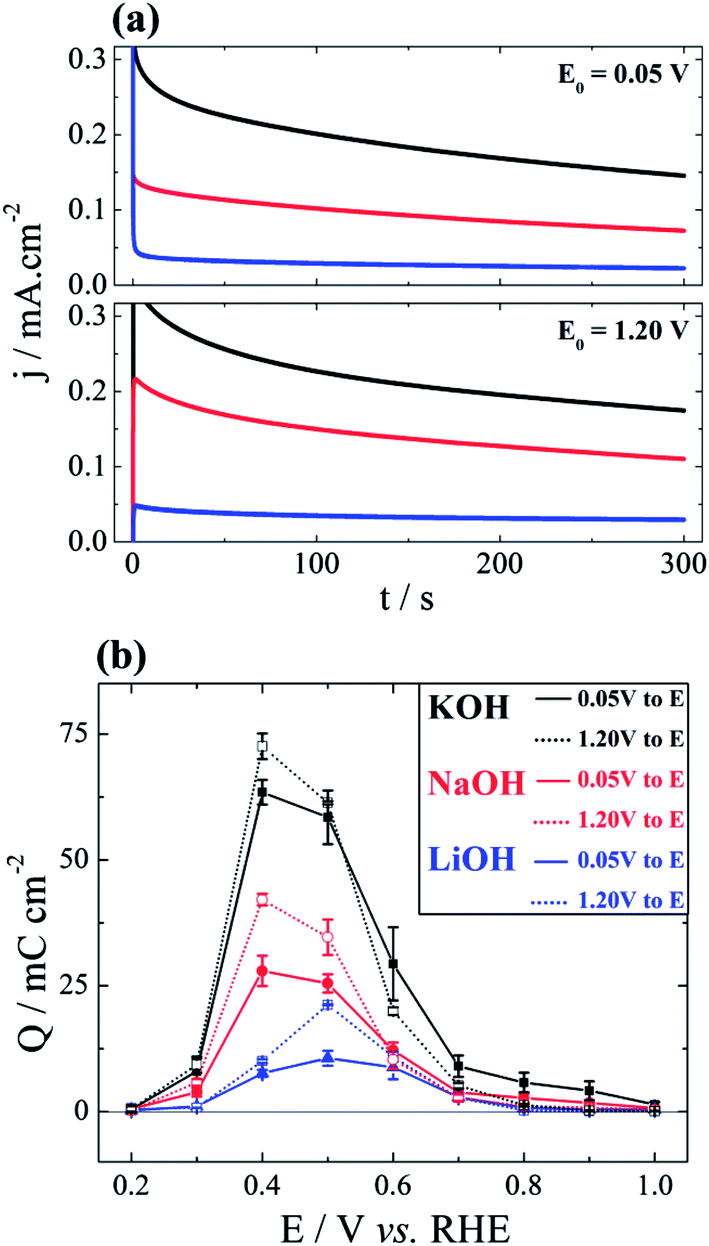 | ||
| Fig. 3 Charges of chronoamperometric experiments. (a) chronoamperometry at E = 0.40 V. E0 refers to polarization pre-measurement. (b) Comparison between two distinct pre-treatments. Full-line: polarization in 0.05 V for 15 s and step to target potential. Dotted-line: polarization in 1.2 V for 15 s and step to target potential. | ||
When compared to the voltammetric profiles provided in Fig. 1(b), it turns out to be clearer in Fig. 3(b) that the cation inhibition becomes less important as the potential increases. Similarly to that discussed above for the base-CV, this is due to the weaker interaction between cations and positively charged surface. Although expected, this trend becomes even more transparent here because of the three discernible oxidation peaks. Finally, it is also noteworthy that the maximum activity, irrespective to the initial potential, occurs at 0.40 V for solutions containing Na+ and K+, and at 0.50 V for Li+. This effect further evidences the dependence interfacial concentration of OHad–M+(H2O)x clusters on the nature of alkali cation and on the electrode potential.
There is an increasingly interest in the electro-oxidation of small organic molecules in alkaline media mainly motivated by the recent development in alkaline fuel cells.13,18 The capital role exerted by the nature of the alkali metal cation present in the electrolyte has been demonstrated for a number of systems.1–6,19 The intricate dependence of cation blockage on the electrode potential evidenced here for this relatively simple model-system expands the knowledge on this relatively recently discovered phenomena. As further evidenced by some intriguing kinetic aspects found under oscillatory regime in alkaline media and not yet understood,20–23 there are still key questions to be answered from fundamental perspective to allow rational design of practical systems.
4. Conclusions
The electrocatalytic oxidation of formate ions on polycrystalline platinum was investigated in the presence of different alkali metal cations. The activity was found to strongly depend on the cation dissolved in the electrolyte and it generally increases in the sequence: Li+ < Na+ < K+. This trend has been found in other systems and it has been attributed to the surface blockage caused by non-covalent interactions between hydrated alkali metal cations and adsorbed oxygenated species on platinum. Peculiar to the electro-oxidation of formate ions, however, was the intricate inhibition caused by the broad and multi-peaked current-potential features. In particular, the use of smaller cations promoted a splitting of one oxidation peak at intermediate potentials and probably anticipated the oxidation of highly stable adsorbate.Acknowledgements
EGM, BAP and HV acknowledge São Paulo Research Foundation (FAPESP) for financial support (grants #2009/07629-6, #2012/07313-1, #2012/24152-1, and #2012/21204-0). HV (#306151/2010-3) acknowledges Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for financial support.References
- C. Stoffelsma, P. Rodriguez, G. Garcia, N. Garcia-Araez, D. Strmcnik, N. M. Marković and M. T. M. Koper, J. Am. Chem. Soc., 2010, 132, 16127 CrossRef CAS PubMed.
- E. Sitta, B. C. Batista and H. Varela, Chem. Commun., 2011, 47, 3775 RSC.
- C. A. Angelucci, H. Varela, G. Tremiliosi-Filho and J. F. Gomes, Electrochem. Commun., 2013, 33, 10 CrossRef CAS.
- D. Strmcnik, K. Kodama, D. Van Der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, Nat. Chem., 2009, 1, 466 CrossRef CAS PubMed.
- D. Strmcnik, D. F. van der Vliet, K. C. Chang, V. Komanicky, K. Kodama, H. You, V. R. Stamenkovic and N. M. Marković, J. Phys. Chem. Lett., 2011, 2, 2733 CrossRef CAS.
- I. Katsounaros and K. J. Mayrhofer, Chem. Commun., 2012, 48, 6660 RSC.
- J. Jiang, J. Scott and A. Wieckowski, Electrochim. Acta, 2013, 104, 124 CrossRef CAS.
- J. Joo, T. Uchida, A. Cuesta, M. T. Koper and M. Osawa, J. Am. Chem. Soc., 2013, 135, 9991 CrossRef CAS PubMed.
- P. A. Christensen and D. Linares-Moya, J. Phys. Chem. C, 2010, 114, 1094 CAS.
- J. John, H. Wang, E. D. Rus and H. D. Abruña, J. Phys. Chem. C, 2012, 116, 5810 CAS.
- R. A. Munson, J. Electrochem. Soc., 1964, 111, 372 CrossRef CAS.
- P. A. Christensen, A. Hamnett and D. Linares-Moya, Phys. Chem. Chem. Phys., 2011, 13, 11739 RSC.
- E. Antolini and E. R. Gonzalez, J. Power Sources, 2010, 195, 3431 CrossRef CAS.
- A. M. Bartrom, J. Ta, T. Q. Nguyen, J. Her, A. Donovan and J. L. Haan, J. Power Sources, 2013, 229, 234 CrossRef CAS.
- Y. Y. Gao, C. H. Tan, Y. P. Li, J. Guo and S. Y. Zhang, Int. J. Hydrogen Energy, 2012, 37, 3433 CrossRef CAS.
- B. C. Batista and H. Varela, J. Phys. Chem. C, 2010, 114, 18494 CAS.
- C. A. Angelucci, H. Varela, E. Herrero and J. M. Feliu, J. Phys. Chem. C, 2009, 113, 18835 CAS.
- J. Jiang and A. Wieckowski, Electrochem. Commun., 2012, 18, 41 CrossRef CAS.
- B. B. Berkes, G. Inzelt, W. Schuhmann and A. S. Bondarenko, J. Phys. Chem. C, 2012, 116, 10995 CAS.
- C. P. Oliveira, N. V. Lussari, E. Sitta and H. Varela, Electrochim. Acta, 2012, 85, 674 CrossRef CAS.
- E. Sitta, R. Nagao and H. Varela, PLoS One, 2013, 8, e75086 CAS.
- E. Sitta, M. A. Nascimento and H. Varela, Phys. Chem. Chem. Phys., 2010, 12, 15195 RSC.
- E. Sitta and H. Varela, Electrocatalysis, 2010, 1, 19 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |