
Enhanced photodegradation activity of methyl orange over Z-scheme type MoO3–g-C3N4 composite under visible light irradiation†
Abstract
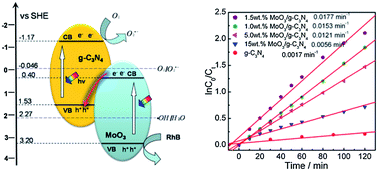
Novel Z-scheme type MoO3–g-C3N4 composites photocatalysts were prepared with a simple mixing–calcination method, and evaluated for their photodegradation activities of methyl orange (MO). The optimized MoO3–g-C3N4 photocatalyst shows a good activity with a kinetic constant of 0.0177 min−1, 10.4 times higher than that of g-C3N4. Controlling various factors (MoO3–g-C3N4 amount, initial MO concentration, and pH value of MO solution) can lead to the enhancement of the photocatalytic activity of the composite. Only MoO3 and g-C3N4 are detected with X-ray diffraction (XRD) and Fourier transform infrared spectroscopy (FT-IR) spectra. N2 adsorption and UV-vis diffuse reflectance spectroscopy (DRS) results suggest that the addition of MoO3 slightly affects the specific surface area and the photoabsorption performance. The transmission electron microscopy (TEM) image of MoO3–g-C3N4 indicates a close contact between MoO3 and g-C3N4, which is beneficial to interparticle electron transfer. The high photocatalytic activity of MoO3–g-C3N4 is mainly attributed to the synergetic effect of MoO3 and g-C3N4 in electron–hole pair separation via the charge migration between the two semiconductors. The charge transfer follows direct Z-scheme mechanism, which is proven by the reactive species trapping experiment and the ˙OH-trapping photoluminescence spectra.
 Please wait while we load your content...
Please wait while we load your content...

