Enhancement of Pt–Ru catalytic activity for catalytic wet air oxidation of methylamine via tuning the Ru surface chemical state and dispersion by Pt addition†
Aiying Songabc and
Gongxuan Lu*a
aState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, P. R. China. E-mail: gxlu@lzb.ac.cn; Fax: +86 931 4968178
bChemical Physics Laboratory of Gansu Provincial Center for Disease Control and Prevention, Lanzhou 730020, P. R. China
cUniversity of Chinese Academy of Science, Beijing 10080, P. R. China
First published on 14th March 2014
Abstract
The enhancement of Pt–Ru catalytic activity for catalytic wet air oxidation (CWAO) of methylamine (MA) was achieved by addition of Pt to Ru catalyst. Pt addition improved the catalytic activity of the bimetallic catalyst due to tuning the Ru surface chemical state and the dispersion of active species. MA was totally mineralized at 200 °C over the Pt–Ru catalyst, while the temperatures required for total mineralization of MA over Pt and Ru catalysts were 240 and 210 °C, respectively. XPS results revealed that addition of Pt could promote the formation of metallic Ru meanwhile TEM and CO chemisorption results confirmed that the addition of Pt promoted the dispersion of active species in the bimetallic catalyst.
Introduction
The release of a great deal of wastewater containing hazardous compounds has caused severe environmental problems. Developing efficient technologies for decomposition of pollutants in wastewater in an environmentally friendly way is thus urgent.1–4 To date, many wastewater treating technologies have been developed, such as biological techniques,5 physico-chemical treatment,6,7 advanced oxidation processes (AOPs), etc. Among them, CWAO has attracted great attention for its powerful capabilities in treatment wastewater containing organic pollutants, which is dilute to be incinerated or very concentrated/toxical to be biologically processed.8–10Originated from WAO technique, CWAO was originally developed by Zimmermann and was first industrialized in the late 1950s.11 By taking advantages of highly reactive oxygen species generated in CWAO process, hazardous compounds can be completely mineralized to CO2, H2O and/or N2 or alternatively to easily biodegradable by-products by controlling reaction conditions.12,13 Compared with WAO, the temperature and time CWAO required for mineralization of hazardous compounds were greatly reduced due to the presence of catalysts.14
Homogeneous catalysts employed in the CWAO are mainly copper, ferric, and manganese salts. These catalysts are quite efficient since they can fully contact with the pollutants at the molecular level.15 However, those catalysts are hard to be recycled. In contrast, the heterogeneous catalysts can be conveniently recovered from the reaction medium by simple filtration. So far, many heterogeneous catalysts have been invented, the active components of these catalysts are mainly transition metal oxides and metallic noble metals, such as Co, Ni, Cr, Cu Pt, Ru, Pd, and so on.16–27 Transition metal oxides are not very selective and stable in CWAO process. Although noble metal-based catalysts are much more expensive than transition metal oxides, their excellent performance and good stability make them receiving much attention in the past decades.11,23,26–28
The pathways and mechanism of CWAO of organic amine have not been fully understood yet. Qin had proposed that the formation of N2 was from the reaction between NOH* and NH*, which were generated during ammonia oxidation process.18 However, Lee indicated that the homogeneous ionic reaction between ammoniac ions and nitrite which was produced by ammonia oxidation was a major route of N2 formation.21,29 Pathways and mechanism of nitrogenous organic compounds can be generally summarized as follows: the cleavage of C–N bond giving carboxyl acid and ammonia, and then these intermediates were further oxidized into CO2 and N2 as well as other by-products.30

In this work, MA is selected as an objective because it is the simplest organic amine contains both C and N atoms, in the meantime, is a common pollutant presenting in the wastewaters of chemical and pharmaceutical industries.31,32 In order to treat such a kind of compound, a stable catalyst is necessary. ZrO2 is used as support because ZrO2 is resistant to acidic and alkaline media, although it has a disadvantage of low specific surface. Alumina has high specific surface area but unstable under the strong acidic and alkaline conditions. Therefore, a composite support Al2O3–ZrO2 were used in the current CWAO process.33 In this paper, Pt–Ru, Pt, and Ru catalysts supported on Al2O3–ZrO2 were prepared by impregnation method and a detail investigation of their catalytic activities in CWAO of MA was performed. The influences of temperature and liquid hourly space velocity (LHSV) on products distribution were investigated, meanwhile, the pathways and mechanism of degradation of MA was discussed.
Experimental
All reagents were of analytical reagent grade. Commercial alumina (>95%, spherical shape, Kaixin Alumina Co. Ltd., China) was crushed to about 25–45 mesh and subsequently calcined at 600 °C for 6 h prior to further use. The alumina particles were modified with zirconia by the impregnation method as follows: 30 g of alumina particles were impregnated into 30 ml aqueous solution containing 5.24 g of Zr(NO3)4·5H2O overnight, and then dried at 120 °C for 12 h followed a calcination treatment at 500 °C for 8 h in air. The composition of the Al2O3–ZrO2 support was measured by XRF. The content of Al2O3 and ZrO2 are about 97.82 and 2.18%, respectively.Pt–Ru/Al2O3–ZrO2, Pt/Al2O3–ZrO2, and Ru/Al2O3–ZrO2 catalysts were also prepared by impregnation method using H2PtCl6·6H2O and RuCl3·xH2O as Pt and Ru precursors, respectively. The total loading amount of metal in three catalysts was fixed at 5 wt% with respect to Al2O3–ZrO2 and for the preparation of Pt–Ru/Al2O3–ZrO2 catalyst the weight ratio of Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru kept at 1:1. Briefly, after impregnation of 10 g of modified alumina particles into 10 ml of precursor solution which contains desired amount of H2PtCl6 and/or RuCl3, the precursor catalysts were dried at 120 °C for 8 h, calcined at 300 °C for 8 h, and finally reduced in H2 flow (40 ml min−1) at 300 °C for another 8 h. The prepared catalysts were denoted as Pt–Ru, Pt, and Ru for simplicity, respectively.
:Ru kept at 1:1. Briefly, after impregnation of 10 g of modified alumina particles into 10 ml of precursor solution which contains desired amount of H2PtCl6 and/or RuCl3, the precursor catalysts were dried at 120 °C for 8 h, calcined at 300 °C for 8 h, and finally reduced in H2 flow (40 ml min−1) at 300 °C for another 8 h. The prepared catalysts were denoted as Pt–Ru, Pt, and Ru for simplicity, respectively.
Catalytic activities of three catalysts were tested in an automated and computer-controlled continuous-flow catalytic evaluation apparatus specialized for heterogenous catalysts (PengXiang Technology Company, Tianjin China), as shown in Fig. 1. Briefly, pre-mixed MA solution (2400 ± 120 ppm) was introduced to the vaporizing chamber by using a peristaltic infusion pump (Lab Alliance Series I, USA), after vaporizing, the mixture of MA and steam were merged with oxygen stream (flow rate: 300 ml min−1) in a T-joint and then were introduced to the reaction tube which was charged with 10 ml of catalysts. The gas and steam, which passed the catalyst bed and flowed out at the bottom of reactor, were cooled with a cold trap and separated in a gas–liquid separator. The temperature and LHSV were set at each experiment at the desired values. The LHSV was defined as LHSV = Fliq/Vcat, h−1; where Fliq = volumetric flow rate of feed solution (ml h−1), and Vcat = catalyst volume (ml). The reaction liquid at the outlet was periodically extracted from the liquid collector and analyzed for TOC, NH4+, NO2−, and NO3− concentrations. Here, Total Organic Carbon (TOC) is defined as the concentration of total organic compound containing carbon atoms except CO2, CO and related substances such as carbonate, bicarbonate. Measurement of TOC can provide a rapid and accurate method of determining the concentration of organic contamination, because the amount of organic carbon in wastewater is an indicator of the organic characteristics of the waste effluent. In our work, the value of TOC of input solution and collected liquid after reaction corresponds to the concentration of methylamine because there were no any other organic carbon-containing intermediates formed during the CWAO of MA according to GC/MS analytical results.
 | ||
| Fig. 1 Schematic diagram for CWAO of MA. | ||
The elemental analysis of the catalysts was conducted by X-ray fluorescence (XRF) on a Netherlands PANalytical spectrometer. X-ray photoelectron spectroscopy (XPS) measurements were performed on K-alpha-surface analysis (Thermon Scientific) using X-ray monochromatization. X-ray diffraction (XRD) patterns of all catalysts were recorded on a Rigaku B/Max-RB diffractometer with a nickel filtrated Cu Kα radiation operated at 40 kV and 40 mA. Transmission electron microscopy (TEM), high-resolution TEM (HRTEM) and elemental mapping images for the catalyst samples were taken by a Tecnai G2-F30 field emission transmission electron microscope operating at accelerating voltage of 300 kV. The specific surface area, total pore volume, and average pore width of the supports and catalysts were determined from the adsorption and desorption isotherms of N2 at −196 °C using a Micromeritics ASAP 2010 instrument. For GC-MS analysis, a gas chromatography-mass spectrometer (GC-MS) (Agilent 7890 A) with a HP-5MS capillary column (30 m × 0.25 mm × 0.25 mm) coupled with an Agilent 5975 mass spectrometer (Agilent Technologies, Palo Alto, CA) was used. TOC was measured using an Analytik Jena Multi N/C 2100 TOC analyzer (Analytik Jena, Germany). Concentrations of ammonia, nitrite and nitrate ions in the collected liquid were determined using colorimetric method according to Chinese national standard methods (GB/T 5750-2006). The selectivity towards nitrogen (N2) was computed via material balance across ‘N’ atom. H2-TPR experiments were carried out by passing a 5% H2 in Ar stream (flow rate: 15 ml min−1) through the catalysts (50 mg). The temperature increased from 50 to 500 °C at a linearly programmed rate of 10 °C min−1. A TCD detector was used to determine the amount of H2 consumed. CO chemisorption was measured with a Micromeritics ChemiSorb 2750 instrument (Micromeritics, USA). All catalysts were reduced in H2 diluted with He (10% v/v) flow at 300 °C for 120 min. After reduction, catalysts were then flushed at 300 °C for 90 min under He to remove physisorbed hydrogen. The catalysts were subsequently cooled under the same He stream. The chemisorbed CO was analyzed at 35 °C.
Results and discussion
The surface chemical states of metal NPs in Pt–Ru, Pt, and Ru catalysts were investigated with XPS technique. The most intensive photoemission lines of Pt 4f7/2 and Ru 3d5/2 levels are overlapped with Al 2p and C 1s lines from alumina support and carbon contaminants, respectively. Therefore the Pt and Ru surface species were investigated by analyzing the Pt 4d5/2 and Ru 3p3/2 lines. Fig. 2 shows the Pt 4d5/2 XPS spectra of Pt–Ru and Pt catalysts. The Pt 4d5/2 peak centered at 315.2 eV in Pt–Ru belongs to Pt0 species while the Pt 4d5/2 peak at 317.0 eV in Pt catalyst is assigned to Pt2+ species.34 The XPS results indicate the presence of Pt metal in the Pt–Ru catalyst while the Pt oxide in the Pt catalyst, which reveals that the Pt precursor can be more easily converted into metallic Pt in the presence of Ru. | ||
| Fig. 2 XPS Pt 4d5/2 spectra for (a) Pt–Ru and (b) Pt catalysts. | ||
The similar results can be observed from the XPS spectra of Ru based catalysts (Fig. 3). The binding energy Ru 3p3/2 (462.4 eV) in Pt–Ru catalyst indicates the presence of metallic Ru while the binding energy Ru 3p3/2 (463.3 eV) in Ru catalyst reveals the presence of Ru oxides.35 This effect is similar to that of Ru-promoted reduction of Pt precursor in Pt–Ru catalyst, suggesting that co-existence of Pt and Ru favours the formation of metallic species in the bimetallic catalyst. The presence of metallic species may contribute to the enhancement of catalytic activity of Pt–Ru for CWAO of MA because the catalytic activity of metallic Pt and Ru are higher than that of their oxides and this issue will be further discussed in our future work.
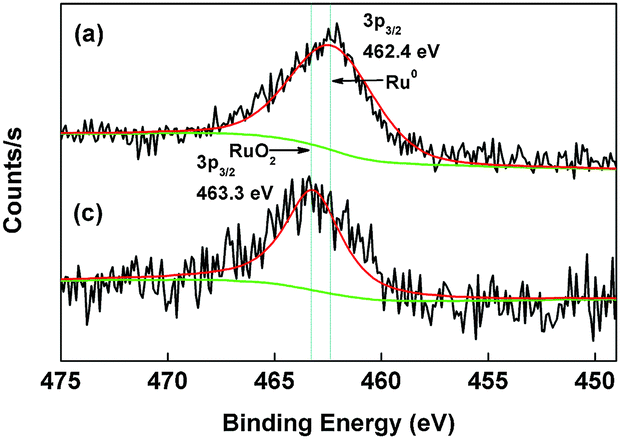 | ||
| Fig. 3 XPS Ru 3p3/2 spectra for (a) Pt–Ru and (c) Ru catalysts. | ||
Fig. 4(a–e) shows X-ray diffraction (XRD) patterns of Al2O3, Al2O3–ZrO2, Pt–Ru, Pt, and Ru catalysts. The peaks in Fig. 4a of patterns at 19.5°, 39.6°, 46.0°, 60.6°, and 67.3° can be assigned to the cubic alumina structures (411), (222), (400), (511), and (440). No obvious diffraction peaks of crystalline ZrO2 can be observed in XRD patterns of Al2O3–ZrO2 (Fig. 4b), which is probably due to either small particle size or low content of ZrO2.
 | ||
| Fig. 4 X-ray diffraction patterns of (a) Al2O3, (b) Al2O3–ZrO2, (c) Pt–Ru, (d) Pt, and (e) Ru. | ||
The crystalline structure of metal in Pt catalyst revealed by XRD pattern shows three typical obvious diffraction peaks at around 40.0°, 46.2°, and 81.3°, which can be indexed to (111), (200), and (311) planes of a face-centered cubic (fcc) structure for Pt (Fig. 4d). On the contrary, in the XRD analysis for Ru catalyst that there is only a diffuse diffraction peak at 2θ = 44.0°, corresponding to (100) plane of a primitive hexagonal close-packed (hcp) crystalline Ru (Fig. 4e).
In case of Pt–Ru catalyst, only typical diffraction peaks at 38.4° and 44.0° corresponding to (101) and (100) planes of hcp crystalline Ru can be observed. Whereas no Pt0 species diffraction peaks appear probably due to the intensities of its reflections are weaker than those of Ru species (Fig. 4c). These analytical results suggest that Pt and Ru components in the bimetallic catalyst did not form alloy and maybe existed as separate bimetal architectures. Similar experimental results had been observed by Zhou et al. during preparation of Pt–Ru/AlOOH catalyst by using H2PtCl6·6H2O and RuCl3·xH2O as precursors by co-impregnation and hydrothermo reduction methods.36
In order to further explore crystalline structures and dispersion of metal NPs over Pt–Ru, Pt, and Ru catalysts, the TEM images were taken. Fig. 5a–c show the low-magnification TEM images. As observed from these images, the supported metal NPs in all catalysts are well-dispersed throughout support substrate with a nearly spherical morphology, however, comparing with Fig. 5a–c, it is clear that the small black metal particles are more uniformly dispersed onto the support in bimetallic catalyst than those in the monometallic ones.
 | ||
| Fig. 5 TEM images of Pt–Ru, Pt, and Ru catalysts a, b, and c. The insets in panels a, b, and c are HRTEM of deposited Pt and/or Ru NPs, respectively. | ||
The HRTEM images of Pt–Ru catalyst (insets in Fig. 5a) shows two set of lattice fingers with d-spacing of 0.224 and 0.204 nm, which can be respectively indexed to (111) plane of fcc Pt and (101) plane of hcp Ru, reconforming that Pt and Ru in bimetallic catalyst exists as separate bimetal architectures. For Pt and Ru catalysts, their HRTEM images (insets in Fig. 5b and c) show that d-spacings of adjacent fringe are 0.224 and 0.204 nm, corresponding to the (111) and (101) crystalline planes of fcc Pt and hcp Ru lattice, respectively. These results are in accordance with the XRD results presented above.
In order to confirm the presence of metal elements, three catalysts were characterized by elemental mapping and energy dispersive X-ray spectrometry (EDX) measurements. As shown in Fig. S1,† the elemental mapping images of three catalysts obviously demonstrate that the distributions of the elements Pt and/or Ru. Clearly, the elemental distribution profiles of Pt and Ru indicate that Pt and Ru NPs are well distributed onto the support. In Fig. S2,† EDX results show the corresponding peaks of C, O, Al, Cu, Zr, Pt, and/or Ru of different catalysts.
H2-TPR is conducted over three catalysts to understand the reduction behaviors of Pt and/or Ru oxides in the calcined catalysts, as shown in Fig. 6. The main reduction peak for Pt catalyst presents at 230 °C, which should be attributed to the reduction of PtOx and/or PtOxCly species on the support, and there is no interaction between those species and support.37 The broad peak of Pt reduction means that Pt particle size distributed in a large range and this speculation can be confirmed by its TEM analysis (Fig. 5b). This phenomenon usually occurred on Pt/Al2O3 catalyst prepared by impregnation, in which the particle size was difficult to be delicately controlled.
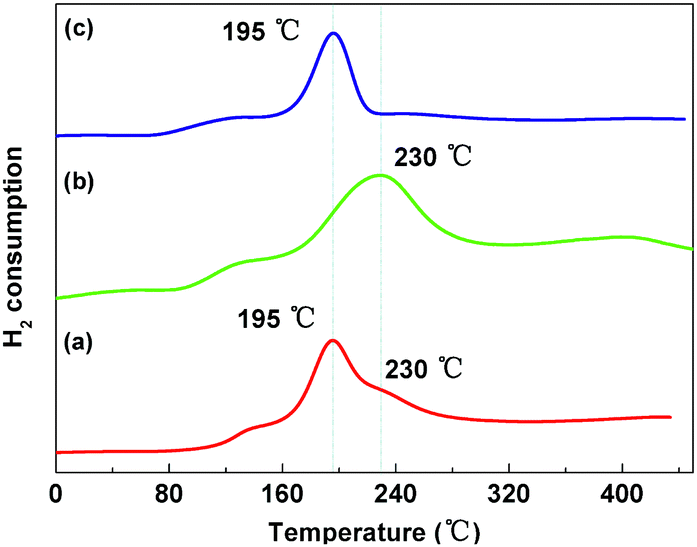 | ||
| Fig. 6 Temperature programmed reduction profiles of (a) Pt–Ru, (b) Pt, and (c) Ru catalysts. | ||
For Ru catalyst, only a sharp reduction peak appears at about 195 °C, which indicates precursor RuCl3 was completely transformed into RuOx after calcination in air.38,39 In case of Pt–Ru catalyst, the peak maximum appears at 195 °C (ascribed to RuOx) with a shoulder at 230 °C (ascribed to PtOx and/or PtOxCly species). TPR results confirm that there is no alloy formation between Pt and Ru.
The surface area, pore volumes, pore sizes of the support, Pt–Ru, Pt, and Ru catalysts with the dispersion of active species in this study are listed in Table 1. The surface area of Al2O3 was hardly changed after modification by ZrO2. But there was a slight loss of surface area for the as-prepared catalysts while the values of the pore volumes and diameters nearly kept constant, which indicated that precious metal NPs were successfully supported onto Al2O3–ZrO2 and the textural properties of support remained unchanged after impregnation. The Pt–Ru and Pt catalysts have dispersions values of 38 and 26%, respectively. However, a poor dispersion (12%) was observed for Ru catalyst. From Table 1 it appears that the combined dispersion of Pt–Ru (38%) matches with the summation of the individual dispersions of Pt (26%) and Ru (12%). This also confirms that there is no interaction between Pt and Ru in the bimetallic catalyst.
| Catalyst | BET (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) | Dispersion (%) |
|---|---|---|---|---|
| Al2O3 | 162 | 0.49 | 12 | — |
| Al2O3–ZrO2 | 163 | 0.42 | 10 | — |
| Pt–Ru | 158 | 0.49 | 12 | 38 |
| Pt | 156 | 0.49 | 13 | 26 |
| Ru | 152 | 0.46 | 12 | 12 |
In order to investigate the effects of reaction temperature on the catalytic activities of Pt–Ru, Pt, and Ru catalysts in CWAO of MA, the TOC (total organic carbon, defined as the concentration of total organic compound containing carbon atoms except CO2, CO and related substances), N-containing compounds yield such as NH4+, NO2−, and NO3− were directly measured and N2 was computed via material balance across ‘N’ atom. TOC conversion (refer to activity) and selectivity of nitrogen species (refer to selectivity) are expressed as the following ratio:
| Activity (%) = (1 − TOCdetermined/TOCini) × 100 |
| Selectivity (%) = (molNlq-class/molMAini) × 100 |
| Selectivity (%) = (2 × molN2/molMAini) × 100 |
Fig. 7 shows the variation of TOC conversion with temperature over Pt–Ru, Pt, and Ru catalysts. It can be seen that reaction temperatures have great influence on catalyst's activity for oxidation of MA. Typically Pt–Ru and Ru catalysts have higher catalytic activity than Pt catalyst for CWAO of MA in examined temperature ranges. For Pt catalyst, MA was completely mineralized until at 240 °C (Fig. 7b), where the TOC conversion reached 100%; however, temperatures required to fully decompose MA were only 200 and 210 °C over Pt–Ru and Ru catalysts (Fig. 7a and c), respectively. These results suggested that Pt catalyst was not very efficient in degradation of MA, though Pt was more active than Ru in ammonia eliminating,1 probably due to Pt has difficulties in cleaving C–N bonds of MA. Pt–Ru catalyst, as expected, demonstrated the highest catalytic activity among three catalysts. Based on XPS and CO chemisorption analysis, it comes to the conclusion that the decrease in temperature for obtaining high activity over Pt–Ru catalyst benefits from the presence of metallic species and the high dispersion of active species in the bimetallic catalyst. In Pt–Ru catalyst, Ru is a primarily active component in C–N bond cleavage for production of NH3 and N2, whereas Pt serves as the promoter to formation of metallic species and enhancement of dispersion of active species in bimetallic catalyst. As a result, Pt–Ru catalyst displayed an excellent activity and the catalytic activity of three catalysts for the CWAO of MA in term of TOC conversion, followed the order of Pt–Ru > Ru > Pt.
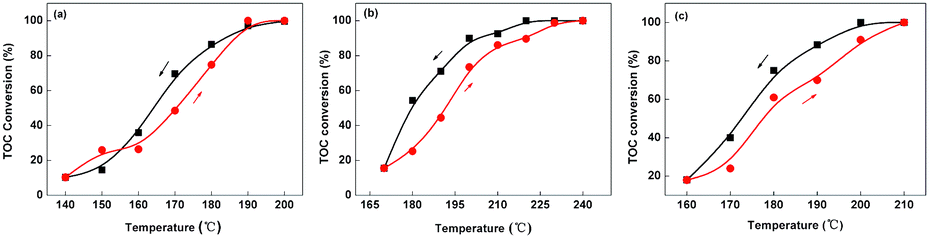 | ||
| Fig. 7 Influence of the temperature on the TOC conversion over (a) Pt–Ru, (b) Pt, and (c) Ru catalysts. | ||
Fig. 8 shows the MA conversion to N2 as a function of temperature over three catalysts. It can be seen from results over Pt–Ru catalyst in Fig. 8 that the C–N bond cleavage only leads to formation of amount of ammoniac ions at low temperature (Fig. S3†) and no N2 produced over Pt–Ru and Pt catalysts. Whereas there were small amounts of N2 still produced over Ru catalyst at low temperature, which indicated that Ru catalyst has excellent selectivity toward N2 in this work. These experimental results revealed that CWAO of MA over three catalysts probably followed two different pathways, that is, over Pt based catalysts, C–N bond of MA was firstly cleaved to form NH3 and then to N2 by further oxidation of NH3, but C–N bond cleavage lead to both NH3 and N2 simultaneously over Ru catalyst. With temperature increasing, ammonia yield reduced after reaching maximum while N2 yields constantly increased and eventually approached to ∼100%. In all experiments, the concentration of NO2− and NO3− are no more than 3.84 and 2.4 ppm, respectively (≤1.6 and ≤1.0‰, as shown in Fig. S4 and S5†).
 | ||
| Fig. 8 Influence of the temperature on the N2 selectivity over (a) Pt–Ru, (b) Pt, and (c) Ru catalysts. | ||
Interestingly, as shown in Fig. 7 and 8, temperature hysteresis can be observed in TOC conversion and selectivity toward N2 over three catalysts. This phenomenon can be also observed in catalytic oxidation of CO over Cu/YSZ (yttria-stabilized zirconia) and Cu/γ-alumina catalysts.40 For example, over Pt–Ru catalyst, the TOC conversion reduced from 87.0 to 69.7% as temperature decreased from 180 to 170 °C, while it was only possible to get the same TOC conversion when temperature increased from 178 to 185 °C during heating direction (Fig. 7a). Based on the hysteresis phenomena presented in Fig. 7 and 8, we tentatively propose that the mechanism of CWAO of MA follows a chemisorption-type mechanism which is similar to that of catalytic oxidation of CO reaction. A possible explanation of hysteresis phenomena observed in this study can be described as following: the temperature, as well known, always controls and determines the adsorption strength of active sites towards reactants. In this experiment, the adsorbed oxygen molecule may not be desorbed from catalysts' surface during cooling until the temperature decreased to a certain one for a long time and the amount of adsorbed oxygen molecules was proportional to concentration of activated oxygen species which directly reacted with MA molecules to mainly produce CO2 and N2, thus TOC conversion and nitrogen selectivity in cooling were higher than those in heating.
Results in Fig. 9, 10 and S6–S8† show that the LHSV greatly influenced TOC conversion and selectivity towards N2, NH4+, NO2−, and NO3− products (refer to N-form products) over three catalyst in examined LHSV range (0.6–5.4 h−1), respectively. As expected, the higher liquid LHSV and lower temperature, the lower conversion of TOC and N2 selectivity (Fig. 9 and 10) were obtained in experiment, indicating the catalytic activities of all catalysts were affected by both temperature and LHSV due to which greatly affect the ratio of MA and activated oxygen species. At high LHSV and low temperature, there was no nitrogen formed and only a large amount of ammonia ions was produced over Pt–Ru catalyst, while both NH4+ and N2 were formed over Ru catalyst (Fig. 10 and S6†). These experimental results reconfirmed that Ru catalyst has better selectivity toward N2 than Pt based catalysts and MA was decomposed according to two different pathways over three catalysts. In addition, high temperature or low LHSV seems to favour formation of nitrite and nitrate by-products (Fig. S7 and S8†).
 | ||
| Fig. 9 Influence of LHSV on the TOC conversion over (a) Pt–Ru, (b) Pt, and (c) Ru catalysts. | ||
 | ||
| Fig. 10 Influence of LHSV on the N2 selectivity over (a) Pt–Ru, (b) Pt, and (c) Ru catalysts. | ||
In order to assess the stability of catalysts, endurance tests were performed at 200, 240, 210 °C and liquid hourly space velocity (h−1, LHSV) of 3.0 h−1 for Pt–Ru, Pt, and Ru catalysts for 300 h, respectively. As a result, the TOC conversion and nitrogen selectivity basically maintained at about 100% during the whole time on stream, which indicates that all catalysts exhibited rather stable catalytic behaviours during 300 h of time on stream.
In order to explore the degradation pathway of MA in the process of catalytic oxidation, GC-MS was applied to identify the hydroxyl amine and organic intermediates in the collected liquid and the condensate. Surprisingly, analytical results show that there were no hydroxyl amine and organic intermediates formed during the CWAO of MA process. On the basis of inorganic product distribution in the collected liquids and the condensate, we proposed a reaction pathway for the degradation of MA on CWAO, as given in Scheme 1. It was concluded that after scission of C–N bond, CH3 fragment was fully converted into CO2 while NH2 fragment was oxidized into N2 and/or NH4+, NO2−, NO3− and NH4+ can be further oxidized into N2, NO2−, and NO3−. It should be noted that CO2 and N2 are the predominant products during MA oxidation reaction.
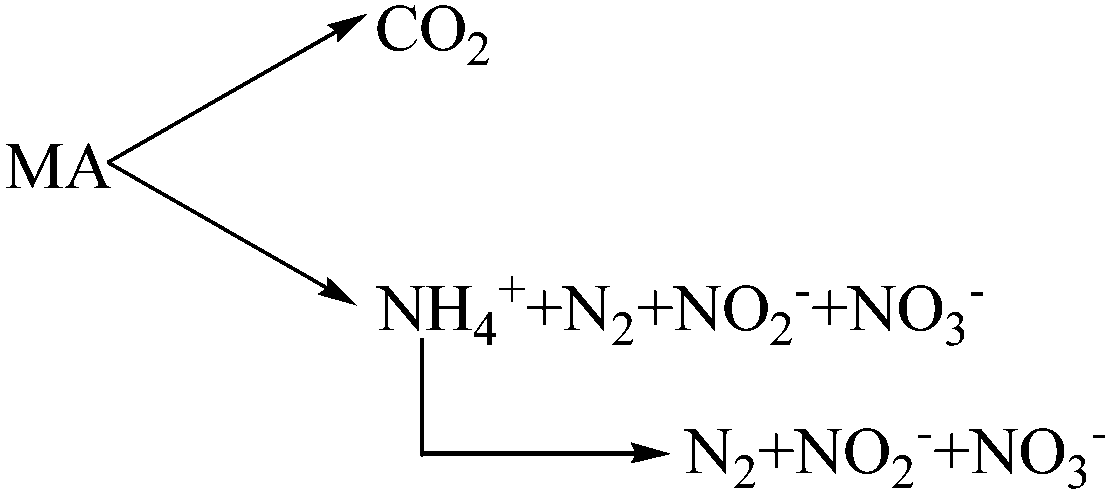 | ||
| Scheme 1 MA oxidation pathways. | ||
Conclusions
XPS studies indicate that the co-existence of Pt and Ru favours the formation of metallic components while TEM and CO chemisorption results confirm the promotion of the dispersion of active species in the bimetallic catalyst, which may contribute to enhancement of catalytic activity of Pt–Ru for CWAO of MA. The TOC conversion and selectivity toward N-form products at different temperatures and LHSV on different catalysts revealed that Pt catalyst was not very efficient in C–N cleavage whereas Ru was capable of both C–N cleavage and ammonia eliminating. Only trace nitrite and nitrate ions were formed during CWAO process, so the experimental procedures employed in this work may provide a useful clue to avoiding formation of these undesired by-products in CWAO of nitrogen-containing compounds.Acknowledgements
We acknowledge the financial supports of the National Science Foundation of China (Grant nos 21373245, 21173242), 973 and 863 Programs of Department of Sciences and Technology of China (Grant nos 2013CB632404, 2012AA051501), and the Project Support of Gansu Provincial Science & Technology Department (1304FKCA085).Notes and references
- J. Barbier, L. Oliviero, B. Renard and D. Duprez, Catal. Today, 2002, 75, 29 CrossRef CAS.
- C. Aguilar, R. Garcia, G. Soto-Garrido and R. Arraigada, Top. Catal., 2005, 33, 201 CrossRef CAS.
- Y. Xu, X. Li, X. Cheng and D. Sun, Environ. Sci. Technol., 2012, 46, 2856 CrossRef CAS PubMed.
- R. Levi, M. Milman, M. V. Landau, A. Brenner and M. Herskowitz, Environ. Sci. Technol., 2008, 42, 5165 CrossRef CAS PubMed.
- C. S. Tripathi and D. G. Allen, Water Res., 1999, 33, 836 CrossRef CAS.
- A. B. Bjerre and E. Soerensen, Ind. Eng. Chem. Res., 1992, 31, 1574 CrossRef CAS.
- C. J. Martino and P. E. Savage, Ind. Eng. Chem. Res., 1997, 36, 1385 CrossRef CAS.
- D. Ghosh and K. G. Bhattacharyya, Appl. Clay Sci., 2002, 20, 295 CrossRef CAS.
- L. Oliviero, J. Barbier and D. Duprez, Appl. Catal., B, 2003, 40, 163 CrossRef CAS.
- E. Castillejos-Lopeza, A. Maroto-Valientea, D. M. Nevskaiaa, V. Munoza, I. Rodríguez-Ramos and A. Guerrero-Ruiz, Catal. Today, 2009, 143, 355 CrossRef.
- F. J. Zimmerman, US Pat. 2, 665, 249, 1950.
- D. K. Lee and D. S. Kim, Catal. Today, 2000, 63, 249 CrossRef CAS.
- C. M. Hung, J. Hazard. Mater., 2010, 180, 561 CrossRef CAS PubMed.
- H. T. Gomes, B. F. Machado, A. Ribeiro, I. Moreira, M. Rosario, A. M. T. Silva, J. L. Figueiredo and J. L. Fari, J. Hazard. Mater., 2008, 159, 420 CrossRef CAS PubMed.
- F. Arena, C. Italiano, A. Raneri and C. Saja, Appl. Catal., B, 2010, 99, 321 CrossRef CAS.
- N. Okada, Y. Nakanishi and Y. Harada, US Pat. 4, 141, 828, 1977.
- H. T. Gomes, P. Selvam, S. E. Dapurkar, J. L. Figueiredo and J. L. Faria, Microporous Mesoporous Mater., 2005, 86, 287 CrossRef CAS.
- J. Qin and K. Aika, Appl. Catal., B, 1998, 16, 261 CrossRef CAS.
- S. K. Kim and S. K. Ihm, Top. Catal., 2005, 33, 171 CrossRef CAS.
- J. Taguchi and T. Okuhara, Appl. Catal., A, 2000, 194–195, 89 CrossRef CAS.
- D. K. Lee, Environ. Sci. Technol., 2003, 37, 5745 CrossRef CAS PubMed.
- S. Kaewpuang-Ngam, K. Inazu and K. I. Aika, Res. Chem. Intermed., 2002, 28, 471 CrossRef CAS.
- G. Sun, A. Xu, Y. He, M. Yang, H. Du and C. Sun, J. Hazard. Mater., 2008, 156, 420 Search PubMed.
- N. D. Tran, M. Besson, C. Descorme, K. Fajerwerg and C. Louis, Catal. Commun., 2011, 16, 98 CrossRef CAS.
- F. Nunez, D. Gloria, F. Tzompantzi and J. Navarrete, Ind. Eng. Chem. Res., 2011, 50, 2495 CrossRef CAS.
- J. Barbier, Jr, L. Oliviero, B. Renard and D. Duprez, Top. Catal., 2005, 33, 77 CrossRef.
- S. Imamura, I. Fukuda and S. Ishida, Ind. Eng. Chem. Res., 1988, 27, 718 CrossRef CAS.
- D. Lee, J. Cho and W. Yoon, Chemosphere, 2005, 61, 573 CrossRef CAS PubMed.
- G. R. Reddy and V. V. Mahajani, Ind. Eng. Chem. Res., 2005, 44, 7320 CrossRef CAS.
- N. Grosjean, C. Descorme and M. Besson, Appl. Catal., B, 2010, 97, 276 CrossRef CAS.
- M. Abalos, J. M. Bayona and F. Ventura, Anal. Chem., 1999, 71, 3531 CrossRef CAS PubMed.
- F. Sacher, S. Lenz and H. J. Brauch, J. Chromatogr. A, 1997, 764, 85 CrossRef CAS.
- F. Núñez, G. D. Angel, F. Tzompantzi and J. Navarrete, Ind. Eng. Chem. Res., 2011, 50, 2495 CrossRef.
- G. Corro, J. L. G. Fierro and V. C. Odilon, Catal. Commun., 2003, 4, 371 CrossRef CAS.
- A. S. Aricò, V. Baglio, A. Di Blasi, E. Modica, P. L. Antonucci and V. Antonucci, J. Electroanal. Chem., 2003, 557, 167 CrossRef.
- Y. F. Zhou, H. Y. Fu, R. X. Li, H. Chen and X. J. Li, Catal. Commun., 2009, 11, 137 CrossRef CAS.
- H. C. Yao and M. Sieg, J. Catal., 1979, 59, 365 CrossRef CAS.
- P. G. J. Koopman, A. P. G. Kieboom and H. V. Bekkum, J. Catal., 1981, 69, 172 CrossRef CAS.
- P. Betancourt, A. Rives, R. Hubaut, C. E. Scott and J. Goldwasser, J. Appl. Catal. A, 1998, 170, 307 CrossRef CAS.
- W. P. Dow, Y. P. Wang and T. J. Huang, J. Catal., 1996, 160, 171 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra00646a |
| This journal is © The Royal Society of Chemistry 2014 |