A green route to synthesize poly(lactic acid)-based macromonomers in scCO2 for biodegradable nanoparticle production†
Raffaele Ferraria,
Claudio Maria Pecorarob,
Giuseppe Stortic and
Davide Moscatelli*a
aDepartment of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Via Luigi Mancinelli 7, Milano, 20131, Italy. E-mail: davide.moscatelli@polimi.it
bDipartimento di Ingegneria Chimica, Gestionale, Informatica e Meccanica, University of Palermo, Viale delle Scienze Ed. 6, Palermo, 90128, Italy
cInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, Zurich, 8093, Switzerland
First published on 21st February 2014
Abstract
Poly(lactic acid)-based macromonomers, aimed at biomedical applications and with well-defined average chain length, are produced through catalytic ring-opening polymerization of L,L-lactide co-initiated by a co-monomer bearing a double bond. Reactions have been carried out in supercritical carbon dioxide (scCO2) at different temperatures, ranging from 90 to 130 °C. The resulting oligomers have been characterized by different techniques (1H-NMR, 13CNMR, MALDI-TOF, ESI, GPC, FT-IR, TGA), which show that oligomers with narrower molecular weight distribution are produced at the lowest temperature. In addition, a significant reduction of the impact of the secondary reactions has been found at this same temperature (90 °C), leading to a crude product with enhanced purity. Finally, the synthesized macromonomers have been used to produce NPs whose degradation behaviour has been investigated. An improved control of the degradation rate is observed for the NPs synthesized using the macromonomer produced in scCO2 at 90 °C.
1. Introduction
Poly(lactic acid) (PLA) is a biodegradable and biocompatible polyester with a wide spectrum of applications, ranging from packaging to tissue engineering and invasive drug delivery.1–3 Poly(lactic acid) (PLA) can be produced by direct polycondensation of lactic acid4,5 or ring-opening polymerization (ROP) of lactide (LT),1,6 the cyclic dimer of lactic acid. The latter route is industrially applied to produce high molecular weight polymers.7 PLA mechanical properties are strongly affected even by small amounts of residual monomer as well as of by-products coming from secondary reactions.Literature studies show that several side reactions take place when producing polyesters at high temperature (higher than 130 °C), being the cyclization of the growing PLA chains the dominant mechanism.8 Other relevant reactions are intermolecular and intramolecular transesterifications which are responsible of different side products and broaden the molecular weight distribution (MWD).9,10 Aimed to reduce the impact of such reactions, low operating temperature should be used for bulk polymerization, even though such choice is strongly limited by the high melting point of LT (95–99 °C). To reduce the reaction temperature, organic solvents can be added to the reaction mixture, thus introducing additional issues of separation and purification of the final product. As an alternative, higher temperatures (around 180 °C) are applied in combination with a careful control of the maximum residence time, since these side reactions are usually slower than propagation reaction.11
Supercritical carbon dioxide (scCO2) is nowadays largely used in post-production treatments of PLA, usually aimed to purification or production of blended polymers with unique properties.12,13 Due to its solvent power scCO2 represents an attractive medium for polymerization reactions alternative to conventional organic solvents.14,15 Moreover, due to its remarkable plasticization ability when dissolved into polymers, lower reaction temperatures can be accessed when running the reaction in scCO2. Finally, the possibility to completely remove the solvent by depressurization provides an additional major advantage when biomedical applications are considered. As a matter of fact, several recent studies have been published, mainly focused on the comparative evaluation of PLA produced in bulk and in scCO2.16–20 In particular, the possibility of improving the polymer characteristics by reducing the reaction temperature was evaluated only by Blakey and co-workers who performed the reaction at low temperature in presence of an organo-catalyst.19
Recently, we developed a strategy to produce nanoparticles (NPs) made of degradable poly-ε-caprolactone based on macromonomers prepared through ROP of ε-caprolactone co-initiated by 2-hydroxyethylmethacrylate (HEMA).21 As an evolution of this approach, the production of PLA-based NPs using LT for the macromonomer synthesis was explored.22 In particular, such macromonomers are made of short and degradable PLA chains with well-defined number of lactic acid units (from 2 to 15) linked to an HEMA molecule which provides the vinyl end group. Such macromonomers (hereinafter HEMA-LAn) have been recently polymerized in emulsion to produce NPs for biomedical applications.23 The resulting polymer chains are composed of a poly(HEMA) backbone where each repeating unit is grafted with n units of PLA; notably, the degradation rate of the produced NPs can be easily tuned by changing n.24 With this respect the synthesis of macromonomers with low and well-controlled molecular weight is highly desirable in order to increase the NP degradation rate. Moreover, side products that can affect the emulsion polymerization step and the final polymer properties, such as species with double vinyl groups that can lead to cross-linked polymers, should be strictly avoided.
Since the feasibility of producing low and medium molecular weight PLA at low temperature in scCO2 has been already proved,17 the same approach is exploited in this work to synthesize low molecular weight macromonomers. Namely, HEMA-LAn macromonomers with n equal to 6 and 8 have been produced at different temperatures using tin octoate as catalyst. The presence of scCO2 allows the melting of LT thus allowing to substantially decrease the reaction temperature without introducing possible issues due to the solvent removal. 1H-NMR has been used to characterize such macromonomers and obtain an optimum reaction time in scCO2. TGA and FT-IR have been adopted to confirm their synthesis, while MALDI-TOF and ESI, highly detailed characterization techniques for low molecular weight polymers, have been used to investigate the relevance of secondary products; in this way, different species present in the final polymer at different reaction conditions could be identified, along with the corresponding side reactions.25,26 GPC analyses have been also performed and combined with MALDI-TOF data giving a first estimation of Mark–Houwink parameters for such macromonomers. The improved purity and the narrower MWD of the oligomers produced in scCO2 at lower temperature has been clearly evidenced. Finally, HEMA-LA8 produced in scCO2 at 90 °C has been used to produce NPs with a reduced content of tin (25 ppm), thus suitable for further bio-applications, in order to confirm the improved applicative behaviour of the material produced in scCO2.24 The degradation behaviour of such particles has been compared to that of NPs obtained from macromonomers synthesized in bulk conditions at 130 °C. In conclusion it has been possible confirm that the different swelling and degradation behaviours of NPs are ascribed to the different characteristics of the initial macromonomers.
2. Experimental section
2.1 Materials
L,L-Lactide (LT, PURAC, 99% purity), 2-hydroxyethylmethacrylate (HEMA, Sigma Aldrich, purity ≥99%) and 2-ethylhexanoic acid tin(II) salt (Sn(Oct)2; Sigma Aldrich, purity ∼95%) were used as received. For gel permeation chromatography (GPC), tetrahydrofuran (THF; Sigma Aldrich, ≥99.7% purity) was used as eluent. CDCl3 (Sigma Aldrich) was used to dissolve samples for 1H-NMR and 13C-NMR analysis. Finally, CH2Cl2 and CHCl3 (both Sigma Aldrich) were used to dissolve samples for MALDI-TOF and ESI. For NPs production, potassium persulfate (KPS, Sigma, ≥99% purity) and sodium dodecyl sulfate (SDS, Merck, purity ∼90%) were used.2.2 Macromonomer synthesis
A thermostated stainless steel reactor previously applied to polymerization reactions carried out in scCO2 was used.27 Desired amounts of monomer, catalyst and initiator were put in a glass vial which was inserted in the reactor. The reactor was sealed and CO2 (99.95% pure, PanGas, Switzerland) was flushed using a high-pressure piston pump (NWA GmbH, Germany) to remove air humidity. Then, the reactor was heated up to the reaction temperature (90, 110 and 130 °C) and pressurized with scCO2 up to 100 bar. The amount of CO2 fed to the reactor was measured using a mass flow meter. 9.45, 10.50, and 13.05 g of CO2 were charged at 130 °C, 110 °C, and 90 °C, respectively; the corresponding absolute error was estimated as ±0.01 g. At the operating conditions, a two phase system was clearly observed: a supercritical CO2-rich phase and a liquid monomer-rich phase. This configuration is fully consistent with the literature for the system LT–CO2 at similar conditions.14,19 Therefore, the reaction was taking place in a liquid mixture and with limited amounts of reactants initially solubilized in the supercritical phase. Since the reaction locus is the liquid, such reactants are transported back to the reacting phase during the process. At the end of the reaction the reactor was vented, opened and the oligomers were removed. The reaction recipes were: 1 g of L,L-lactide, 0.20 g of HEMA and 3.1 mg of Sn(Oct)2 for the synthesis of HEMA-LA8 and 1 g of L,L-lactide, 0.32 g of HEMA and 5.0 mg of Sn(Oct)2 for HEMA-LA6. The amount of Sn(Oct)2, was kept smaller than 50 ppm with respect to LT. The co-monomer HEMA acts as an initiator; the initiator-to-catalyst molar ratio was maintained equal to 1/200. Due to the small amounts of catalyst, very slow reaction rates are established, with reaction times longer than typical literature values.18 To evaluate the conversion evolution in time, different reactions have been performed with different durations; 1, 2, 3 and 4 days at 90 °C; 6, 14, 18 and 24 h at 110 °C; 2, 3, 4, 5 and 6 h at 130 °C. In order to fully remove the unreacted LT, the crude macromonomer was first dissolved in chloroform and dropwise added to an excess of cyclohexane where the purified product was precipitated.282.3 Macromonomer characterization
The macromonomer sampled at different reaction times was characterized via, 1H-NMR. Oligomers obtained at the optimum reaction time (as discussed later) were also characterized through 13C-NMR, Thermo Gravimetric Analysis (TGA), Infrared Spectroscopy (FT-IR), MALDI-TOF, ESI and GPC.Structure and composition of the macromonomers were also determined by 1H-NMR and 13C-NMR in CDCl3 using 500 MHz Ultrashield NMR spectrometer (Bruker, Switzerland). As already reported, the average number molecular weight (Mn) of the macromonomers has been evaluated by 1H-NMR through eqn (1):29
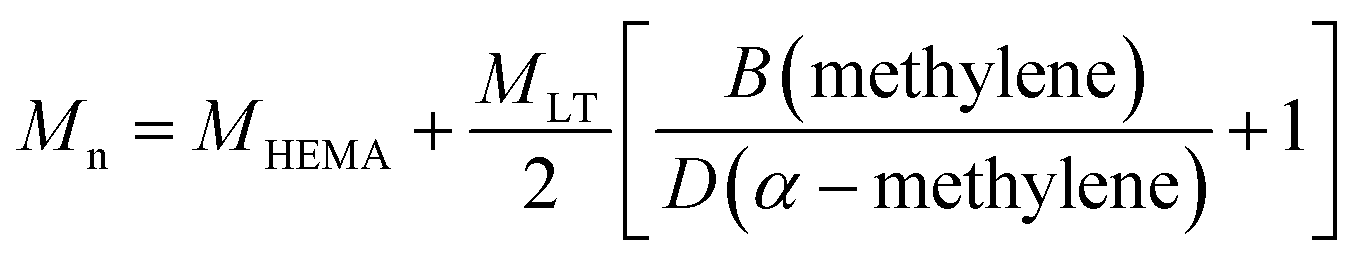 | (1) |
Thermal properties were studied using TGA (STA 6000, Perkin Elmer, US). Samples of 10 mg were analysed under nitrogen (30 mL min−1) from 30 °C to 350 °C at heating rate of 10 °C min−1.
FT-IR spectra were recorded using attenuated total reflection absorbance detection (TENSOR Series FT-IR spectrometer, Bruker, Switzerland). For each sample, scans were recorded between 4000 cm−1 and 500 cm−1 (resolution equal to 8 cm−1).
MALDI-TOF mass spectra were recorded using an Ultraflex II TOF Bruker spectrometer (Bremen, Germany) using 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]-malononitrile (DCTB) as matrix material. Samples co-crystallized with the matrix on the probe were ionized by Smart Bean laser pulse (337 nm) and accelerated under 25 kV with time-delayed extraction before entering the time-of-flight mass spectrometer. Matrix and sample were separately dissolved in dichloromethane and mixed in ratio 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 matrix:sample. To produce some specific adducts, sodium ions are added (1% sodium acetate in methanol). 1 μL mixture of matrix and sample was spread on a MALDI-TOF MS probe and air-dried. All spectra were performed in positive reflection mode; external calibration was performed by using peptide calibration standard II (700–3200 Da) from Care (Bruker, Switzerland).
:1 matrix:sample. To produce some specific adducts, sodium ions are added (1% sodium acetate in methanol). 1 μL mixture of matrix and sample was spread on a MALDI-TOF MS probe and air-dried. All spectra were performed in positive reflection mode; external calibration was performed by using peptide calibration standard II (700–3200 Da) from Care (Bruker, Switzerland).
Macromonomers were also analysed by using a maXis ESI-Q-TOF (Bruker, Switzerland) mass spectrometer equipped with an automatic syringe pump for sample injection (KD Scientific, US). The ESI-Q-TOF mass spectrometer was running at 4500 V, with desolvation temperature of 200 °C. The mass spectrometer was operated in the positive ion mode; nitrogen was used as nebulizer and drying gas. The standard electrospray ion (ESI) source was used to generate ions and chloroform (CH3Cl) was used as a solvent. Sixty shots from each spot were averaged to obtain one mass spectrum. The ESI-Q-TOF MS instrument was calibrated in the m/z range 50–1300 with the use of an external calibration standard (Tunemix solution), supplied by Agilent.
The MWD of all samples were measured by SEC (Agilent, 1100 series, US) equipped with two detectors, ultraviolet (UV) and differential refractive index (RI), using THF as eluent with 1 mL min−1 flow rate and temperature of 35 °C. After the precolumn, two columns in series were used: a PLgel 5lm MIXED-C column (Polymer Laboratories, length: 300 mm; diameter: 7.5 mm; measuring range: 2000–2000000 Da) and an oligopore column (Polymer Laboratories; length: 300 mm; diameter: 7.5 mm: measuring range, 0–4500 Da). Universal calibration based on polystyrene standards (Polymer Laboratories, 162–28500 Da) and Mark–Houwink–Sakurada parameter values for polystyrene measured in THF at 35 °C (K1 = 1.14 × 10−4 dL g−1 and a1 = 0.716)30 were applied as discussed later.
2.4 Nanoparticles synthesis and degradation
NP synthesis, as well as degradation studies were performed as recently described by Yu et al.22 A monomer starved semi-batch polymerization reaction was performed to obtain PLA-based NPs, while their degradation was monitored at constant temperature (50 °C) by dynamic light scattering (DLS, Zetasizer Nano, Malvern, UK).3. Results and discussion
3.1 Preliminary characterization of macromonomers
Macromonomers, typically in the form of transparent and viscous liquids, produced at different reaction times as reported in the Experimental section have been analysed by 1H-NMR. A first characterization of macromonomer synthesis has been done by estimating time evolutions of conversion versus time and number average molecular weight (Mn) versus conversion as shown in Fig. 1 for the different reaction temperatures.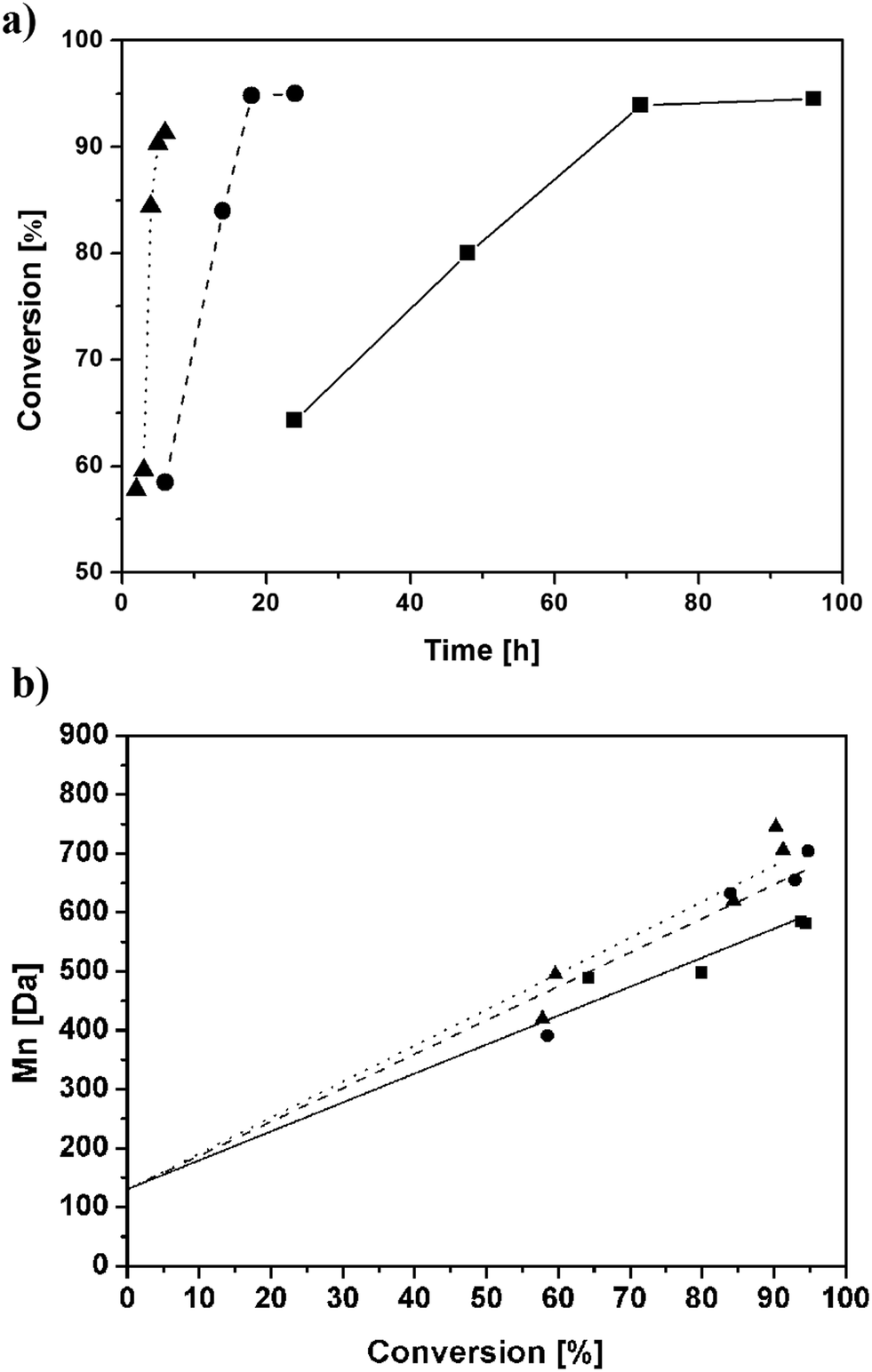 | ||
| Fig. 1 Conversion versus time (a) and Mn versus conversion (b) for HEMA-LA8 produced in scCO2 at 90 °C (■), 110 °C (●), and 130 °C (▲) as evaluated by 1H-NMR. Lines represent data fitting for 90 °C (straight line), 110 °C (dashed line), and 130 °C (dotted line). | ||
Each single experimental value reported in the figure represents an independent reaction; then, in order to evaluate the corresponding relative error, a specific experiment (6 hours of reaction time at 130 °C) was repeated three times at constant operating conditions. The 1H-NMR results show an average error on the average molecular weight smaller than 10% (705, 740, and 748 Da), thus confirming the acceptable reproducibility of the experiments. Then, it is worth mentioning that Mn decreases at very long polymerization time. A decrease of molecular weight has been already reported at high conversion values (higher than 95%) and imputed to side reactions.11 Then, a maximum reaction time, which correspond to a range of final conversion from 92% to 95%, specific for each temperature value was set to prevent such decrease (6 h, 18 h and 72 h at 130 °C, 110 °C, and 90 °C, respectively). Fig. 1b also indicate a polymerization behaviour close to livingness. The next part of the discussion focuses on the characterization of macromonomers produced at these optimum reaction times.
A typical 1H-NMR spectrum of HEMA-LA8 macromonomer is shown in Fig. 2a together with peak assignments, which confirm the macromonomer molecular structure (peak integrations are reported in Fig. S1† of the ESI). The peaks of the residual LT are also shown, since they have been used to calculate the monomer conversion (peaks B′). Moreover, 13C-NMR spectrum reported in Fig. 2b further confirms the molecular structure of the macromonomer.
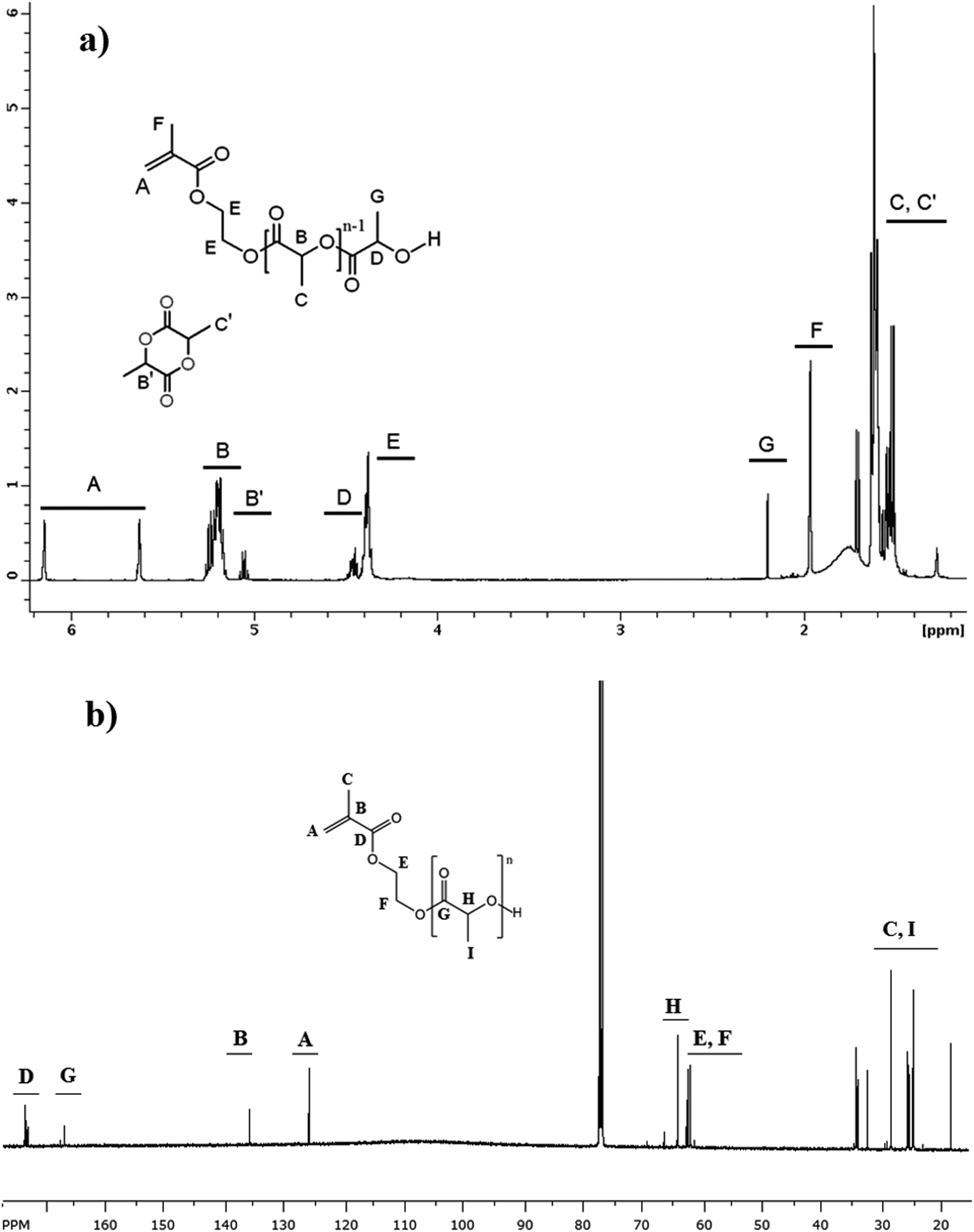 | ||
| Fig. 2 1H-NMR (a) and 13C-NMR (b) spectrum of HEMA-LA8 produced in scCO2 at 110 °C. | ||
As previously shown, the macromonomer formation is confirmed; the calculated values of Mn obtained by 1H-NMR are collected in Table 1.
| Macromonomer | Temp. [°C] | Time [h] | 1H-NMR | |
|---|---|---|---|---|
| n | Mn [Da] | |||
| HEMA-LA8 | 130 | 6 | 8.0 | 705 |
| HEMA-LA8 | 110 | 18 | 8.0 | 704 |
| HEMA-LA8 | 90 | 72 | 6.3 | 581 |
| HEMA-LA6 | 130 | 6 | 5.6 | 536 |
| HEMA-LA6 | 110 | 18 | 4.7 | 465 |
| HEMA-LA6 | 90 | 72 | 4.3 | 440 |
The thermal resistance of the crude macromonomers was then characterized using TGA, heating the polymer from 35 °C to 400 °C. Profiles of thermal decomposition of LT and of two macromonomers recorded at different temperatures are shown in Fig. 3.
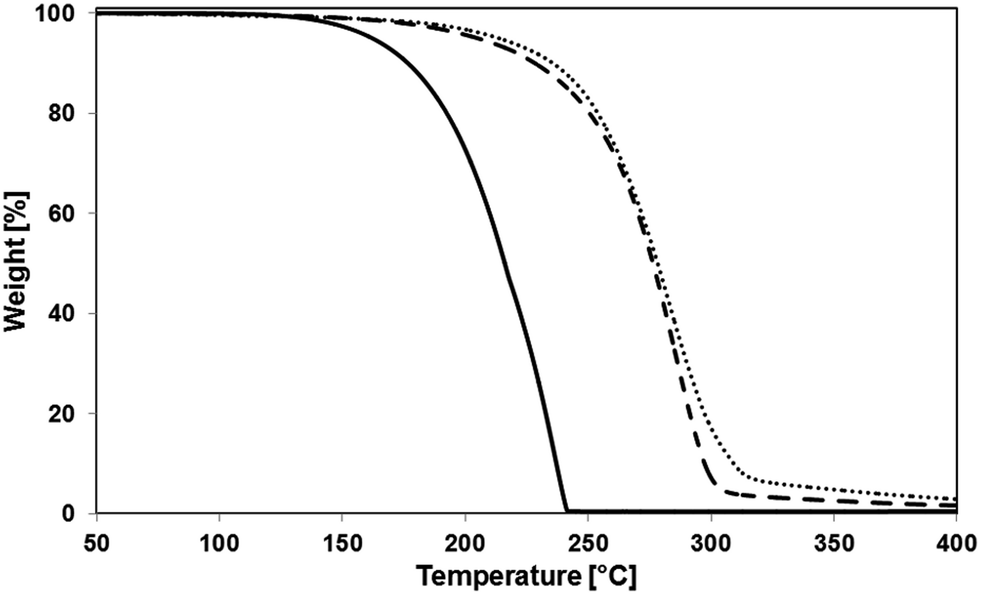 | ||
| Fig. 3 TGA thermograms of L,L-lactide (solid line), HEMA-LA8 produced in scCO2 at 130 °C (dashed line) and at 90 °C (dotted line). | ||
The thermal behaviour of the examined species is quite different: while the thermal degradation of LT is complete at 240 °C, that of HEMA-LA8 just started at the same temperature. The slight weight loss detectable for HEMA-LA8 at temperature below 250 °C is most probably due to the removal of non-reacted LT, while no significant differences are detectable in the two samples produced at different temperatures. The thermal behaviour of HEMA-LA6 is fully comparable to the one observed for HEMA-LA8 (see Fig. S2†).
Finally, FT-IR analysis of the produced macromonomers has been performed to confirm the macromonomer formation and the addition of lactoyl units to the HEMA molecules (detailed results reported in Fig. S3†).
3.2 Role of the side reactions
After the preliminary characterization discussed above, aimed to prove the feasibility of the synthesis of HEMA-LAn macromonomers in scCO2, the detailed characterization of macromonomer molecular structure and purity when synthesized both at lower temperature and suitable reaction time has been performed using MALDI-TOF and ESI. Firstly, MALDI-TOF have been used to evaluate Mn, calculated as 845, 844, and 725 Da for HEMA-LA8, 697, 590 and 581 Da for HEMA-LA6 produced at 130, 100, and 90 °C, respectively. Such average molecular weights from MALDI-TOF are always higher than those measured by GPC and 1H-NMR: this is quite expected, since species at low molecular weight (smaller than 400 Da) are not detected by this technique using the selected calibration. Finally, the polydispersity values (lower than 1.1) confirm the anticipated living behaviour of the reaction.As already reported in the Introduction, side reactions play a relevant role during the ROP of LT at high values of conversion or reaction temperature, even though a non-negligible contribution has been found also at temperature as low as 130 °C.11 Such events lead to MWD broadening, thus decreasing the quality of the final material.28 In particular, in the case of PLA homopolymers, the most important side reactions are inter- and intra-molecular transesterifications, as well as chain scission, and their possible pathways have been summarized in Scheme 1.8,9,32,33 The reaction steps involve both active and dormant PLA chains, as generically indicated by the terminal asterisk.
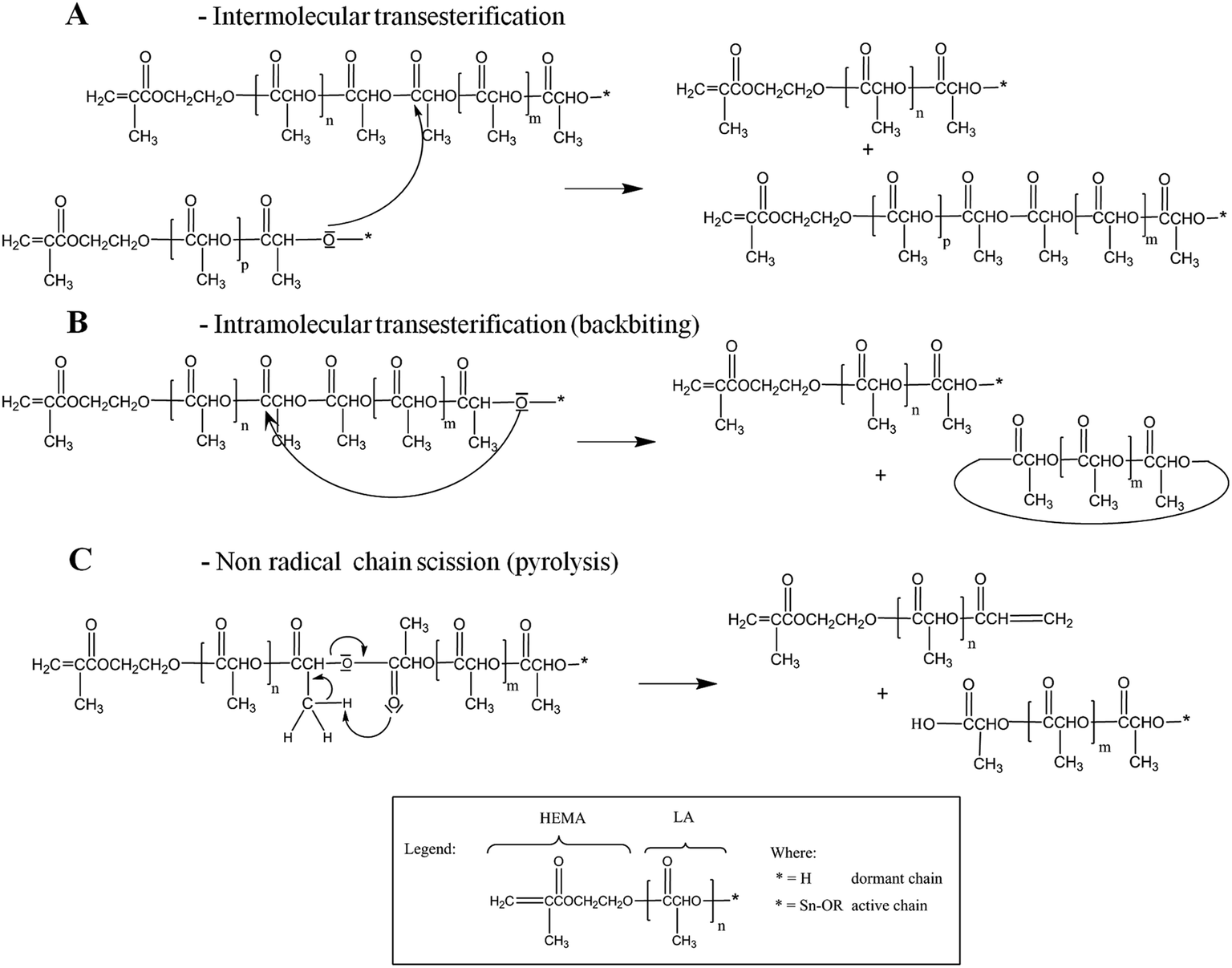 | ||
| Scheme 1 Major secondary reactions involved in the ROP of LT. | ||
Notably, macromonomers with odd number of lactoyl units can be formed only when transesterifications occur, either intermolecular or intramolecular (A and B in Scheme 1). In fact, two lactoyl repeating units are added per each propagating step and the presence of species with an odd number of repeating units can be considered as the proof of the active role of such reactions. In the specific case under examination (HEMA terminal units bearing ester groups), additional transesterification paths become possible, as shown in Scheme 2.
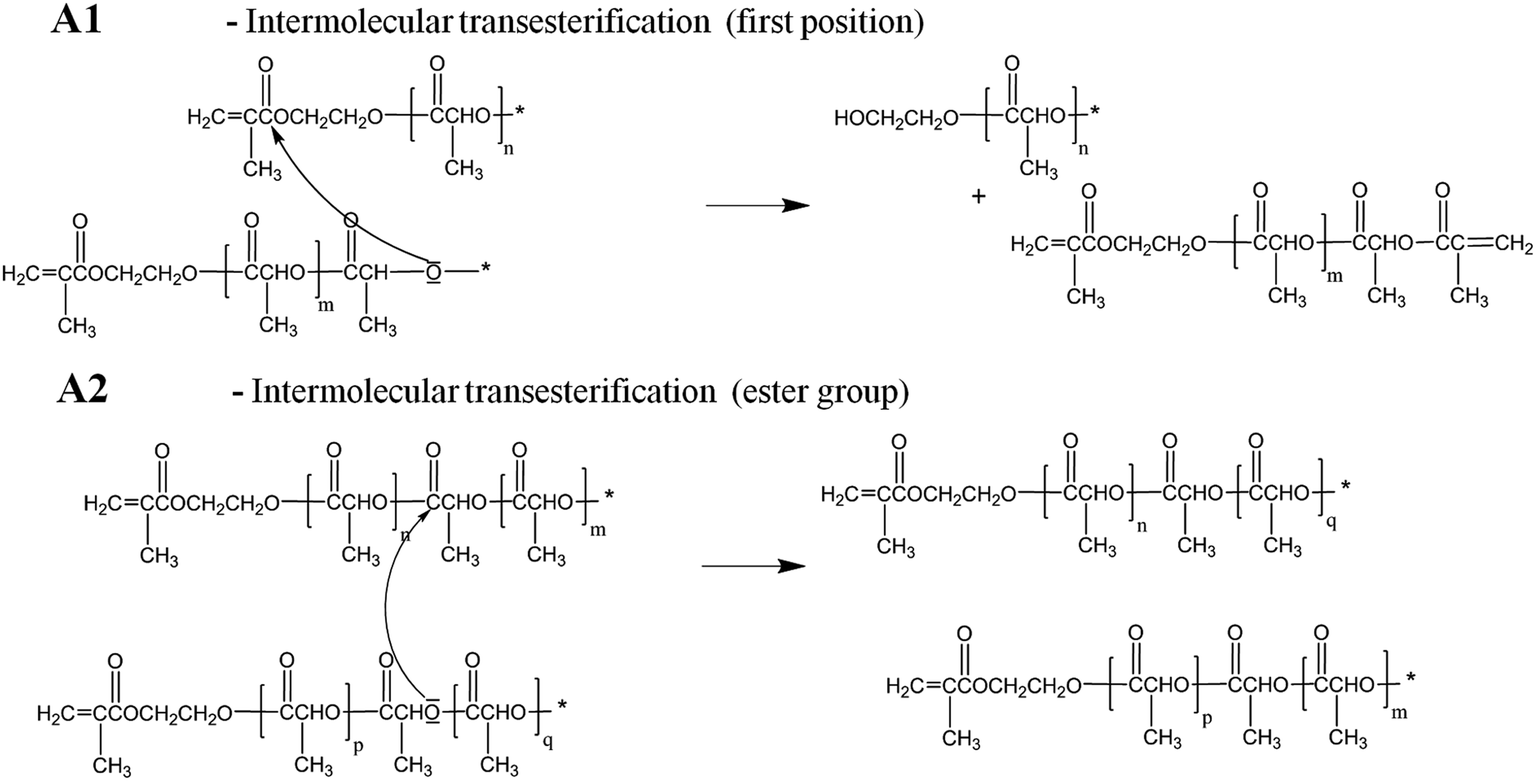 | ||
| Scheme 2 Alternative pathways for transesterification reaction. | ||
Reaction A1, has been previously reported for ε-caprolactone chains initiated by HEMA.34 It is worth mentioning that also ester groups are able to initiate transesterification reactions (A2). Nevertheless, their reactivity is very low compared to that of hydroxyl groups (reaction A in Scheme 1): then, the contribution of reaction A2 has been neglected in this work.35 Finally, reaction C in Scheme 1 is a pyrolysis reaction recently proposed when studying the thermal degradation of PLA.28,36,37
All previous reactions affect the MWD of the oligomers; in particular transesterifications (A and B in Scheme 1) lead to chains with lower or higher number of lactoyl repeating units with respect to the average number of the non-transesterified chains. Moreover, the concentration of small sized cyclic compounds produced in the case of backbiting reaction (B), which happens if the reactive site is an ester group located into the same macromonomer chain, should be minimized since they are unreactive species in the next emulsion polymerization step. Finally, reactions A1 and C, are responsible for the formation of products with two vinyl end groups which could act as crosslinking agents during the following emulsion polymerization step.
In order to assess the effects of the secondary reactions, HEMA-LA8 oligomers produced at 130 °C and 90 °C were analysed through MALDI-TOF. The recorded spectra are shown in Fig. 4 while detailed peak assignments are reported in the ESI (Fig. S4†).
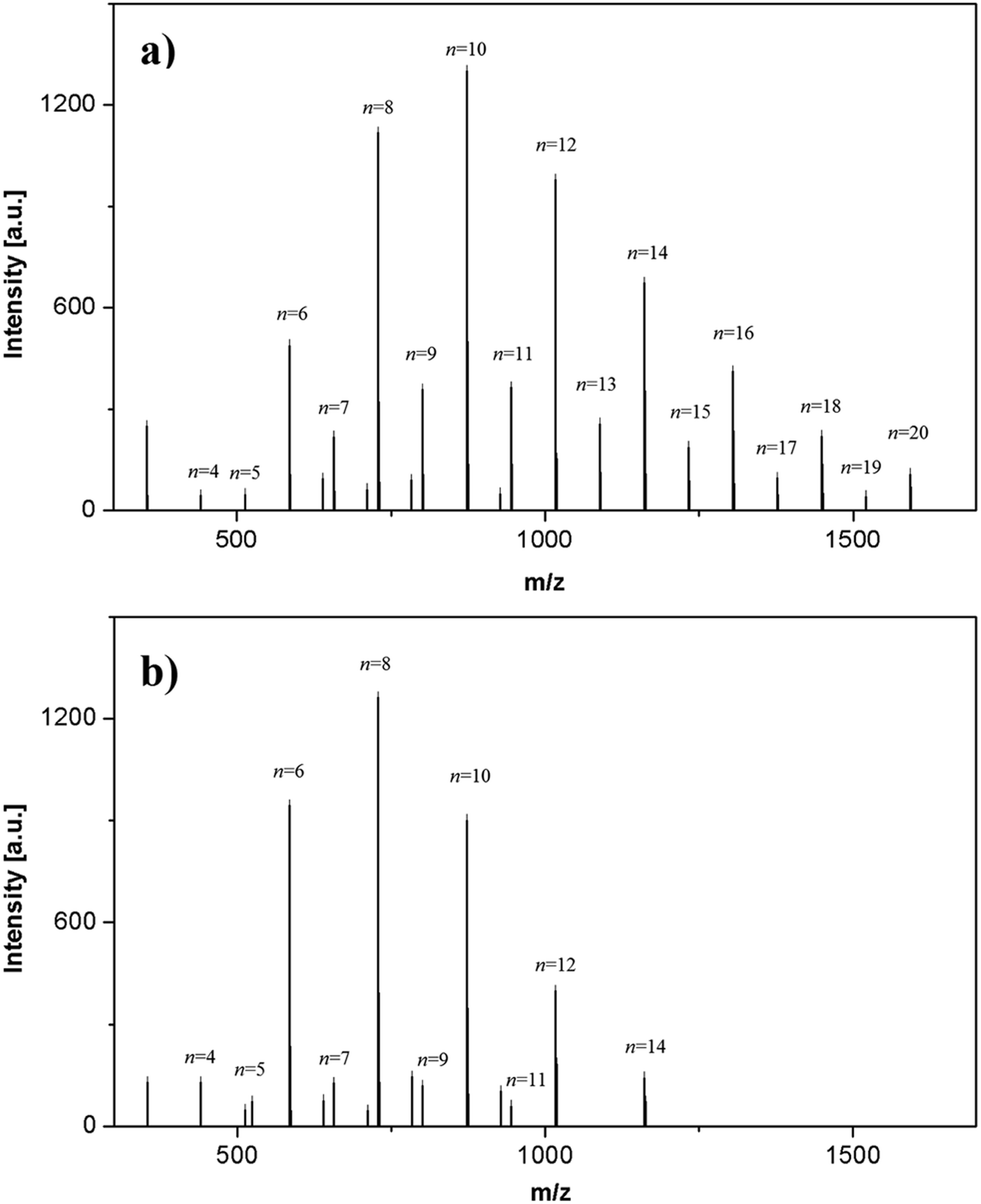 | ||
| Fig. 4 MALDI-TOF of HEMA-LA8 macromonomer produced in scCO2 at 130 °C (a) and 90 °C (b). | ||
The major peaks in the figure correspond to macromonomers of different length associated with the cation Na+. The values of the experimental and theoretical masses, as well as the absolute values of their difference (|Δ(m/z)|), are collected in Table 2: their close agreement confirms the peak assignment. It is important to notice that peaks at very low molecular weight (such as 272 and 353 Da) are due to the matrix.
| Macromonomer (LA units) | m/ztheo [Da] | m/zexp [Da] | |Δ(m/z)| [Da] |
|---|---|---|---|
| 4 | 441.14 | 440.89 | 0.25 |
| 5 | 512.93 | 512.93 | 0 |
| 6 | 585.18 | 584.98 | 0.20 |
| 7 | 657.20 | 657.02 | 0.18 |
| 8 | 729.22 | 729.06 | 0.16 |
| 9 | 801.24 | 801.09 | 0.15 |
| 10 | 873.29 | 873.13 | 0.16 |
| 11 | 945.16 | 945.16 | 0 |
| 12 | 1017.31 | 1017.19 | 0.12 |
| 13 | 1089.33 | 1089.22 | 0.11 |
| 14 | 1161.35 | 1161.24 | 0.11 |
| 15 | 1233.37 | 1233.26 | 0.11 |
| 16 | 1305.39 | 1305.39 | 0.10 |
| 17 | 1377.41 | 1377.30 | 0.11 |
| 18 | 1449.43 | 1449.32 | 0.09 |
| 19 | 1521.45 | 1521.31 | 0.14 |
| 20 | 1593.48 | 1593.35 | 0.13 |
The peaks with the highest intensity in both Fig. 4a and b can be ascribed to macromonomers with even number of lactoyl repeating units. In Fig. 4a two different distributions can be visualized, corresponding to oligomers with even and odd numbers of lactoyl repeating units. On the other hand, oligomers with odd numbers of units are detected at much lower relative intensity in Fig. 4b. Finally, small amount of chains initiated by water are observed in both the products.
The dominant species is the target macromonomer (HEMA-LA8) only when the reaction is carried out at low temperature, while HEMA-LA10 becomes dominant if the reaction is performed at higher temperature. In the latter case, the final MWD is broader than that obtained at 90 °C, meaning that much longer chains than the target value (HEMA-LA8 in this case) are formed. Indeed, macromonomers bearing up to 20 lactoyl units (1593 Da) are detected by MALDI-TOF, while macromonomers with molecular weight higher than 1161 Da (HEMA-LA14) are not detectable when the reaction is performed at 90 °C.
In order to further deepen the characterization of the final products, the relative areas of macromonomer peaks have been compared; since the oligomers possess the same end groups, their ionization energies are comparable, thus allowing to evaluate the relative intensities of the m/z signal to achieve a quantitative evaluation of macromonomer amounts.31,38,39 Then, to estimate the relevance of the transesterification reactions, we calculated the amounts of high molecular weight macromonomers with odd number of repeating units, those which can be produced only by these side reactions. The percentages of peak area of macromonomers with n equal to or higher than 14 are 29%, 14% and 5% when the reaction is performed at 130 °C, 110 °C and 90 °C, respectively. Repeating this evaluation for the entire range of molecular weights but focusing on macromonomers with odd number of lactoyl repeating units, the following percentages are evaluated: 18%, 12%, and 4.6% at 130 °C, 110 °C and 90 °C, respectively. Similar results have been observed for the HEMA-LA6 macromonomer (Fig. S5†). It becomes clear that the transesterification reactions broads the MWD and shifts the molecular weight of the oligomers to higher values when the ROP is performed at 110 or 130 °C. Conversely, no significant broadening is observed if the reaction is carried out at milder temperature, even if the reaction time is much longer (from hours to days), thus proving that side reactions are significantly reduced. It is worth mentioning that the only additional peaks detected non related to the macromonomers correspond to PLA oligomers initiated by water instead than by HEMA: such additional initiator, present as impurity in the raw LT, can be removed through recrystallization in extra-dry toluene,11 a treatment not applied here.
With reference to Scheme 1 no evidences about the presence of the reactives pathway A1 and C were obtained by MALDI-TOF analyses. Then, ESI analyses of the same crude products were performed, since this technique has higher resolution power than MALDI-TOF for very low molecular weight (<2000 Da),40 though not effective for quantitative estimations. A comparison between the ESI spectra of the macromonomers produced at 130 °C and 90 °C is presented in Fig. 5 (detailed peaks analysis is reported in Fig. S6†).
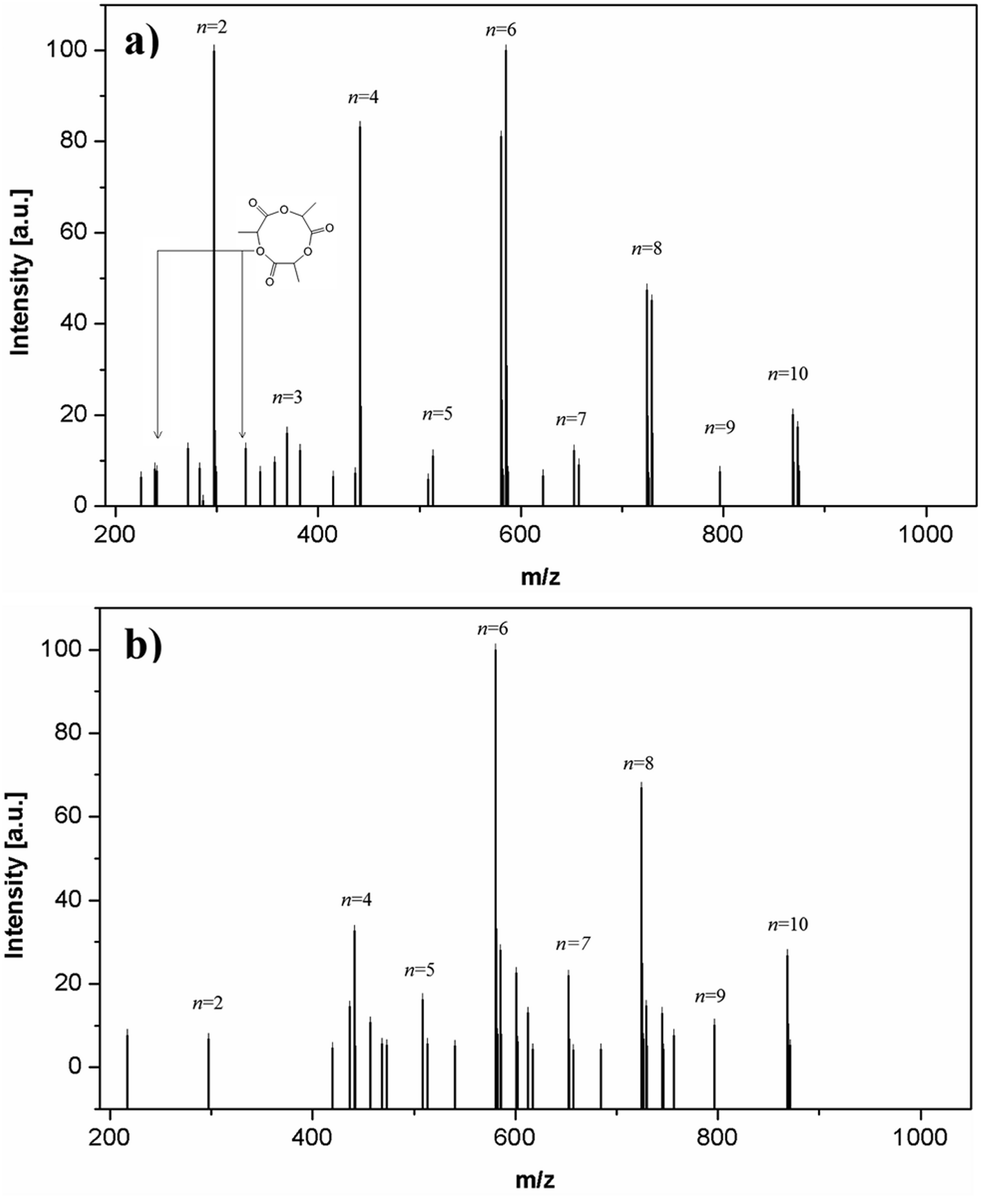 | ||
| Fig. 5 ESI of HEMA-LA8 macromonomer produced in scCO2 at 130 °C (a) and 90 °C (b). | ||
No significant differences were detected in comparison with the MALDI-TOF results in the “high” molecular weight portion of the spectrum (>400 Da), since only species related to macromonomers (associated with cations Na+ or NH4+) and their isotopes were found at both operating temperatures. Conversely, if shorter chains are considered (<400 Da), some relevant differences between macromonomers produced at different temperatures appear. First, PLA macrocycles composed by 3 and 5 lactoyl units were recognized (239.02 and 382.28 Da) at high reaction temperature. These species can be produced only by backbiting reactions, thus confirming the relevance of intramolecular transesterification at 130 °C. Furthermore, chains made of pure PLA ended by an acidic group were also detected and, as previously reported for MALDI-TOF, they were attributed to species initiated by water. Finally, macrocycles were not detected in the macromonomer mixture produced at 90 °C (detailed peaks reported in Fig. S6b†), thus confirming the negligible role of intramolecular transesterifications at this temperature. Since no side products coming from pyrolysis or transesterification to the first position were detected by both MALDI-TOF and ESI, it can be concluded that these reactive pathways are fully negligible in the investigated temperature interval.
Finally, results obtained from MALDI-TOF spectra have been coupled with GPC data to provide a first estimation of Mark–Houwink (MH) parameters for HEMA-LAn. The measured MWD of HEMA-LA8 produced in scCO2 at different temperatures are shown in Fig. 6 as measured by GPC.
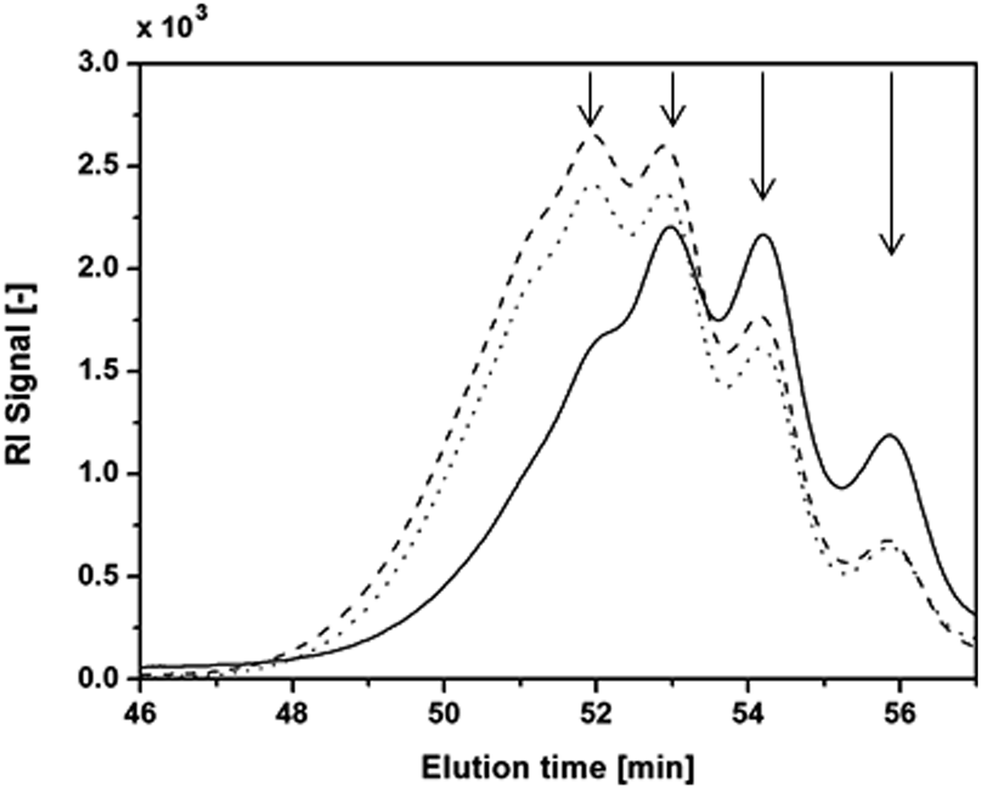 | ||
| Fig. 6 GPC elution curves of HEMA-LA8 produced in scCO2 at 90 °C (solid line), 110 °C (dashed line) and 130 °C (dotted line). | ||
Four main peaks can be identified in all the eluted curves shown in Fig. 6, as indicated by the arrows in the figure. GPC calibrated with PS standards has been adopted to determine the MW correspondent to the four peaks highlighted in the figure, estimated as 1254, 1065, 819, and 517 Da. By comparing these peaks with the relatives areas obtained from MALDI-TOF they have been identified as macromonomers with n equal to 10, 8, 6, and 4, respectively. Then, by applying MH equation, two parameters, a and K, have been obtained leading to obtain universal calibration for HEMA-LAn. These values, reported in Table 3 are quite different from PLA values while they are closer to HEMA-CLn values estimated from viscosity analyses.21
3.3 NP degradation behaviour
Aimed to evaluate the quality of the materials obtained at milder temperature, HEMA-LA6 and HEMA-LA8 produced at 90 °C were finally used to produce NPs through a monomer starved, semibatch emulsion polymerization.22 NP degradation behaviour was compared to that of NPs obtained using the same macromonomers produced at 130 °C in bulk. The size of the NPs produced using macromonomers synthesized at 90 °C are summarized in Table 4. As it can be observed a nice agreement has been found with macromonomers produced at 130 °C as.| n | Reaction medium | Particle size [nm] | Polydispersity index42 [-] |
|---|---|---|---|
| 6 | scCO2 | 28.4 | 0.043 |
| 6 | Bulk | 26.5 | 0.055 |
| 8 | scCO2 | 27.9 | 0.094 |
| 8 | Bulk | 25.3 | 0.055 |
The degradation behaviour of these NPs at 50 °C is shown in Fig. 7 where the evolution of particle size versus time is reported. The figure shows an increase in the particle size of all the selected NPs up to their complete disruption and the formation of a transparent solution where NPs are no longer detected by DLS.
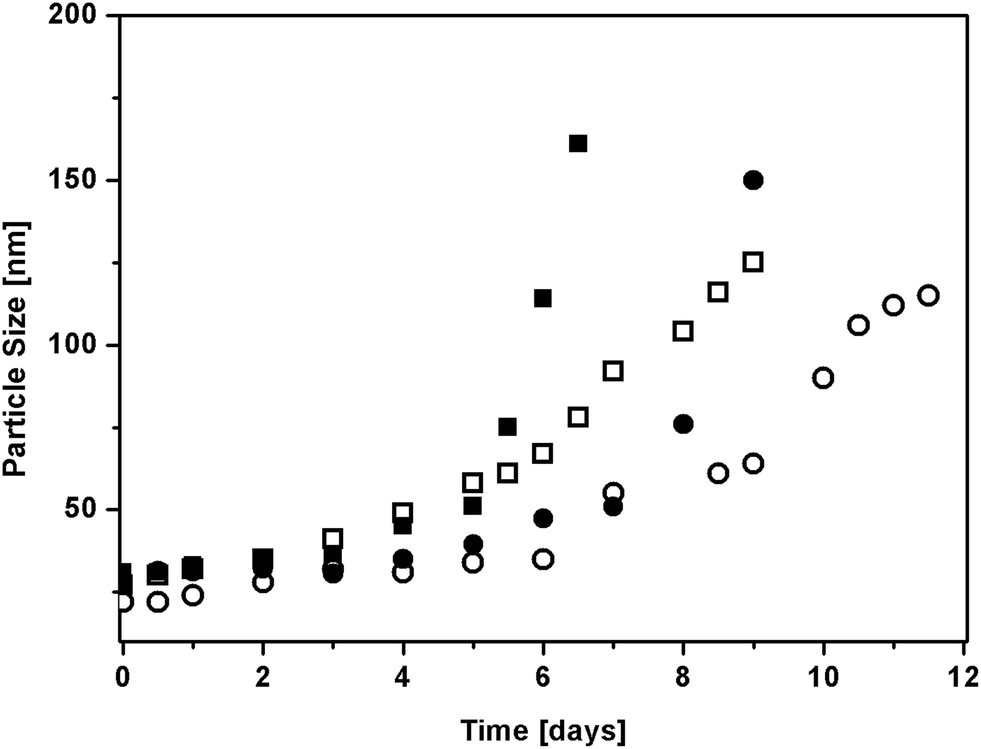 | ||
| Fig. 7 Evolution of particle size vs. time during the degradation of NPs from macromonomers produced in bulk at 130 °C (HEMA-LA6 (□), and HEMA-LA8 (○)) and produced in scCO2 at 90 °C (HEMA-LA6 (■), and HEMA-LA8 (●)). | ||
As reported in the literature by Yu et al.,21 this behaviour was explained by a swelling mechanism: during degradation, the lipophilicity of the NPs decreases because of the reduction of molecular weight due to the elimination of lactic acid and its oligomers due to ester hydrolysis. As a result, water diffuses into the particles increasing their size by swelling as well as further enhancing hydrolysis of PLA side units. Eventually, poly-HEMA chains are formed, whose solubility in water explains particle disappearance. Since longer times are required to hydrolyze longer chains, particle disruption takes place at different times: such times increase at increasing length of the PLA side chains, from 6 days for HEMA-LA6 to 9 days for HEMA-LA8, Therefore, NPs with tunable degradation time are easily produced by tuning the macromonomer size.
Notably, NPs based on HEMA-LA8 synthesized at 90 °C in scCO2 exhibit higher values of the maximum swelling; in fact, their size grows up to about 150 nm before the disruption takes place, which is indeed higher than the final size of the NPs produced with the macromonomer synthesized in bulk. Moreover, their degradation is completed in 9 days, while 12 days are required by NPs based on HEMA-LA8 produced at 130 °C, and the time evolution is clearly different in the two cases. A fully equivalent behaviour was observed for NPs based on HEMA-LA6. Such reduced time of full degradation along with the faster degradation rate can be imputed to the different MWD of the original macromonomers, containing lower fraction of species at high molecular weight when synthesized at higher temperature.
4. Conclusion
PLA-HEMA copolymers with controlled average chain length have been produced from L,L-lactide, using HEMA as initiator and Sn(Oct)2 as catalyst. HEMA-LAn macromonomers with n = 6 and 8 have been produced in scCO2 at constant pressure (100 bar) and different temperatures, from 90 to 130 °C. At each reaction temperature, maximum reaction durations were defined to minimize the impact of side reactions. The produced oligomers were fully characterized by 1H-NMR, TGA, FT-IR, MALDI-TOF, and ESI. MALDI-TOF proved the increased purity (less by-product content) of the macromonomer produced at 90 °C, while the formation of macrocycles by backbiting reactions, non-detectable by MALDI-TOF, was identified by ESI. Such by-products are found when ROP is performed at higher temperature while their amounts becomes negligible if the reaction is carried out at milder temperature. Additional GPC analyses have been used to give a first estimation of MH parameters for HEMA-LAn. The produced macromonomers have been used to synthesize NPs by monomer starved, semi-batch emulsion polymerization. Their degradation rates, tunable by tuning the target PLA chain length, have been compared to that of NPs synthesized using macromonomers produced in bulk at 130 °C. The different degradation behaviours confirm the impact of the reaction conditions applied during the macromonomer synthesis on the properties of the final NPs.Acknowledgements
The authors gratefully acknowledge Louis Bertschi (ETH Zurich, Switzerland) for MALDI-TOF and ESI analyses and Simone Gelosa (Politecnico di Milano, Italy) for TGA analyses.References
- L. T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS PubMed.
- S. Honarbakhsh and B. Pourdeyhimi, J. Mater. Sci., 2011, 46, 2874–2881 CrossRef CAS.
- J. K. Oh, Soft Matter, 2011, 7, 5096–5108 RSC.
- Y. M. Harshe, G. Storti, M. Morbidelli, S. Gelosa and D. Moscatelli, Macromol. React. Eng., 2007, 1, 611–621 CrossRef CAS.
- F. Achmad, K. Yamane, S. Quan and T. Kokugan, Chem. Eng. J., 2009, 151, 342–350 CrossRef CAS PubMed.
- S. Dutta, W. C. Hung, B. H. Huang and C. C. Lin, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Kunkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, Springer-Verlag Berlin, Berlin, 2012, vol. 245, pp. 219–283 Search PubMed.
- X. Pang, X. Zhuang, Z. Tang and X. Chen, Biotechnol. J., 2010, 5, 1125–1136 CrossRef CAS PubMed.
- S. Penczek, R. Szymanski, A. Duda and J. Baran, Macromol. Symp., 2003, 201, 261–269 CrossRef CAS.
- J. Baran, A. Duda, A. Kowalski, R. Szymanski and S. Penczek, Macromol. Rapid Commun., 1997, 18, 325–333 CrossRef CAS.
- A. Kowalski, J. Libiszowski, K. Majerska, A. Duda and S. Penczek, Polymer, 2007, 48, 3952–3960 CrossRef CAS PubMed.
- Y. C. Yu, G. Storti and M. Morbidelli, Macromolecules, 2009, 42, 8187–8197 CrossRef CAS.
- C. A. Kelly, A. Naylor, L. Illum, K. M. Shakesheff and S. M. Howdle, Adv. Funct. Mater., 2012, 22, 1684–1691 CrossRef CAS.
- A. Vega-Gonzalez, P. Subra-Paternault, A. M. Lopez-Periago, C. A. Garcia-Gonzalez and C. Domingo, Eur. Polym. J., 2008, 44, 1081–1094 CrossRef CAS PubMed.
- J. Gregorowicz, J. Supercrit. Fluids, 2008, 46, 105–111 CrossRef CAS PubMed.
- H. Y. Tai, C. E. Upton, L. J. White, R. Pini, G. Storti, M. Mazzotti, K. M. Shakesheff and S. M. Howdle, Polymer, 2010, 51, 1425–1431 CrossRef CAS PubMed.
- R. Mazarro, A. de Lucas, L. I. Cabezas, I. Gracia and J. F. Rodriguez, Macromol. Symp., 2010, 287, 111–118 CrossRef CAS.
- D. Bratton, M. Brown and S. M. Howdle, Macromolecules, 2003, 36, 5908–5911 CrossRef CAS.
- S. Y. Lee, P. Valtchev and F. Dehghani, Green Chem., 2012, 14, 1357–1366 RSC.
- I. Blakey, A. Yu, S. M. Howdle, A. K. Whittaker and K. J. Thurecht, Green Chem., 2011, 13, 2032–2037 RSC.
- D. D. Hile and M. V. Pishko, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 562–570 CrossRef CAS.
- R. Ferrari, Y. C. Yu, M. Morbidelli, R. A. Hutchinson and D. Moscatelli, Macromolecules, 2011, 44, 9205–9212 CrossRef CAS.
- Y. Yu, R. Ferrari, M. Lattuada, G. Storti, M. Morbidelli and D. Moscatelli, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5191–5200 CrossRef CAS.
- R. Ferrari, Y. Yu, M. Lattuada, G. Storti, M. Morbidelli and D. Moscatelli, Macromol. Chem. Phys., 2012, 213, 2012–2018 CrossRef CAS.
- R. Ferrari, C. Colombo, M. Dossi and D. Moscatelli, Macromol. Mater. Eng., 2012, 298, 730–739 CrossRef.
- J. De Winter, V. Lemaur, R. Ballivian, F. Chirot, O. Coulembier, R. Antoine, J. Lemoine, J. Cornil, P. Dubois, P. Dugourd and P. Gerbaux, Chem.–Eur. J., 2011, 17, 9738–9745 CrossRef CAS PubMed.
- I. Osaka, M. Watanabe, M. Takama, M. Murakami and R. Arakawa, J. Mass Spectrom., 2006, 41, 1369–1377 CrossRef CAS PubMed.
- L. I. Costa, G. Storti, M. Morbidelli, X. Zhang, B. Zhang, E. Kasëmi and A. D. Schlüter, Macromolecules, 2011, 44, 4038–4048 CrossRef CAS.
- Y. C. Yu, G. Storti and M. Morbidelli, Ind. Eng. Chem. Res., 2011, 50, 7927–7940 CrossRef CAS.
- R. Ferrari, A. Cingolani and D. Moscatelli, Macromol. Symp., 2013, 324, 107–113 CrossRef CAS.
- K. Liang, M. Dossi, D. Moscatelli and R. A. Hutchinson, Macromolecules, 2009, 42, 7736–7744 CrossRef CAS.
- G. Montaudo, M. S. Montaudo, C. Puglisi, F. Samperi, N. Spassky, A. LeBorgne and M. Wisniewski, Macromolecules, 1996, 29, 6461–6465 CrossRef CAS.
- O. Wachsen, K. H. Reichert, R. P. Kruger, H. Much and G. Schulz, Polym. Degrad. Stab., 1997, 55, 225–231 CrossRef CAS.
- Y. Liu, R. Q. Wei, J. Wei and X. N. Liu, Prog. Chem., 2008, 20, 1588–1594 CAS.
- A. Gallardo, A. Marcos-Fernandez, S. Egri, R. Lebron and E. Piskin, Int. J. Polym. Anal. Charact., 2008, 13, 83–94 CrossRef CAS.
- F. Codari, S. Lazzari, M. Soos, G. Storti, M. Morbidelli and D. Moscatelli, Polym. Degrad. Stab., 2012, 97, 2460–2466 CrossRef CAS PubMed.
- Y. M. Wang, B. Steinhoff, C. Brinkmann and I. Alig, Polymer, 2008, 49, 1257–1265 CrossRef CAS PubMed.
- Y. J. Fan, H. Nishida, Y. Shirai and T. Endo, Polym. Degrad. Stab., 2004, 84, 143–149 CrossRef CAS PubMed.
- Y. J. Li, J. N. Hoskins, S. G. Sreerama, M. A. Grayson and S. M. Grayson, J. Mass Spectrom., 2010, 45, 587–611 CAS.
- L. H. Cohen and A. I. Gusev, Anal. Bioanal. Chem., 2002, 373, 571–586 CrossRef CAS.
- G. Hart-Smith, M. Lammens, F. E. Du Prez, M. Guilhaus and C. Barner-Kowollik, Polymer, 2009, 50, 1986–2000 CrossRef CAS PubMed.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS.
- L. M. Gugliotta, G. S. Stegmayer, L. A. Clementi, V. D. G. Gonzalez, R. J. Minari, J. R. Leiza and J. R. Vega, Part. Part. Syst. Charact., 2009, 26, 41–52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra00333k |
| This journal is © The Royal Society of Chemistry 2014 |