
Revisiting the principles of preparing aqueous quantum dots for biological applications: the effects of surface ligands on the physicochemical properties of quantum dots†
Abstract
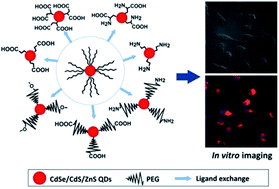
Surface functionalization of quantum dots (QDs) is one of the most important aspects for the design and preparation of the desired QDs for specific biomedical applications. The surface ligands not only render the QDs water-dispersible, but also endow them with different functional groups for bioconjugation. More importantly, as the surface ligand layer on the QD surface is responsible for interacting with the biological environments, the type of surface ligand will greatly affect the response from the cells, such as the cellular uptake and cytotoxicity. In this paper, we investigate the effects of the surface ligand on the physicochemical properties of QDs and examine different QD formulations for possible biomedical applications. Seven types of QD formulations were prepared by anchoring the CdSe/CdS/ZnS QDs surface with either short-chain mercapto ligands (MPA, MSA, cysteine, AET) or PEG derivative ligands (mPEG-SH, CM-PEG-SH, NH2-PEG-SH). We then conducted a systematic study to evaluate the colloidal stability, photostability, cellular uptake and in vitro toxicity of the formulations. The colloidal stability was evaluated by the particle size change in water, acidic/neutral/alkaline buffer solutions and cell culture medium. Our results show that the carboxyl-terminated QDs have the best colloidal stability in water and alkaline solutions. PEG-capped QDs are more stable than short-chain ligand modified QDs in cell culture medium. For the photostability of different QD formulations under UV irradiation, we observed that the MPA-, MSA- and Cys-QDs had better photostability than that of the PEG modified QDs, whereas the AET-QD is the least stable one. Cellular uptake of QDs was evaluated using cell imaging and quantified by flow cytometry. The PEG chains and surface charge of QDs were found to play critical roles in the cellular uptake. Using RAW246.7 macrophage cells as the cellular uptake model, we discovered that the anionic QDs had a much higher uptake compared to the cationic QD formulations. In general, each set of prepared QD formulation with a specific type of surface ligand display certain strengths and limitations in different aspects of their physicochemical properties. Therefore, one should carefully consider and choose the type of QD formulation in the experiments thereby minimizing its impacts arising from their limitations.
 Please wait while we load your content...
Please wait while we load your content...

