
FRET-reporter nanoparticles to monitor redox-induced intracellular delivery of active compounds†
Abstract
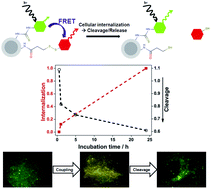
Nanoparticle-mediated drug delivery holds great promise for more specific and efficient therapies, mostly due to their high payload and the integration of targeted delivery functions. However, it is typically not possible to monitor the intracellular release of active compounds directly, which hampers the assessment of novel delivery platforms and non-cytotoxic compounds. Herein, we implemented a FRET (fluorescence resonance energy transfer)-reporter system to semi-quantitatively follow the time-course of the intracellular release of a redox-cleavable compound from a nanoparticle delivery platform. We used silica core–shell particles that could be readily modified with a high density of reactive amino groups, and attached fluorescent reporter molecules as model drug cargo. We coupled a FRET-donor fluorophore via a stable covalent bond, while the FRET-acceptor fluorophore was linked via a disulfide-bridge. Therefore, a loss of FRET reported on redox-induced acceptor compound release. These FRET-reporter nanoparticles allowed us to determine both the time course of cellular internalization as well as the intracellular compound release. Our data show that particle internalization is the rate-limiting step, while compound cleavage occurs quickly after internalization. The presented FRET-reporter approach has the advantage to directly monitor the compound cleavage, as compared to indirect read-outs that are based on their cytotoxic action. We therefore propose that our FRET-reporter particles are suitable to monitor intracellularly redox-releasable compounds that are typically not cytotoxic, such as siRNAs.
 Please wait while we load your content...
Please wait while we load your content...

