
Ultrafine PDMS fibers: preparation from in situ curing-electrospinning and mechanical characterization†
Abstract
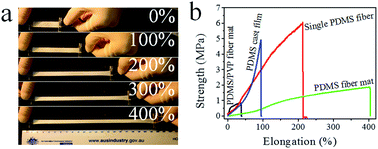
Polydimethylsiloxane (PDMS) fibers with unexpected elasticity were prepared by a modified core–shell electrospinning method using a commercially-available liquid PDMS precursor (Sylgard 184) and polyvinylpyrrolidone (PVP) as core and sheath materials, respectively. The liquid PDMS precursor was crosslinked in situ to form a solid core when the newly-electrospun core–sheath nanofibers were deposited onto a hot-plate electrode collector. After dissolving the PVP sheath layer off the fibers, net PDMS fibers showed larger average diameter than core–sheath fibers, with an average diameter around 1.35 μm. The tensile properties of both single fibers and fibrous mats were measured. Single PDMS fibers had a tensile strength and elongation at break of 6.0 MPa and 212%, respectively, which were higher than those of PDMS cast film (4.9 MPa, 93%). The PDMS fiber mat had larger elongation at break than the single PDMS fibers, which can be drawn up to 403% their original length. Cyclic loading tests indicated a Mullin effect on the PDMS fiber mats. Such a superior elastic feature was attributed to the PDMS molecular orientation within fibers and the randomly-orientated fibrous structure. Highly-elastic, ultrafine PDMS fibers may find applications in strain sensors, biomedical engineering, wound healing, filtration, catalysis, and functional textiles.
 Please wait while we load your content...
Please wait while we load your content...

