
Recent developments in chemistry and biology of curcumin analogues
Abstract
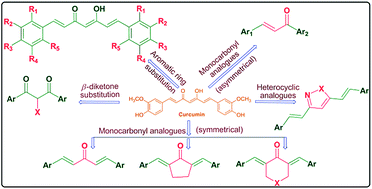
Curcumin, a prominent constituent of the rhizome of Curcuma longa L., possesses versatile biological properties, which is evidenced from the extensive research during the last half century. Curcumin has been shown to exhibit antioxidant, anti-inflammatory, antiviral, antibacterial, antifungal, and anticancer activities and thus has potential against various malignant diseases, such as allergies, arthritis, Alzheimer's disease, and other chronic illnesses. In the last decade it has been much explored and various synthetic analogues have been prepared and evaluated for various pharmacological activities that render it as a lead molecule against several biological targets. To accelerate this lead molecule from kitchen to clinic, around 65 clinical trials are underway worldwide to assess its therapeutic potential. Thus, there is continued interest in the synthesis of new curcumin analogues with a similar safety profile, but increased activity and improved oral bioavailability. The present review article describes recent developments in curcumin chemistry with emphasis on the semi-synthesis, synthesis pharmacological properties and SAR of various curcumin analogues reported from 1994 to mid 2013.
- This article is part of the themed collection: Towards understanding and treating Alzheimer’s disease
 Please wait while we load your content...
Please wait while we load your content...

