
Modeling of chemical–mechanical couplings in anode-supported solid oxide fuel cells and reliability analysis
Abstract
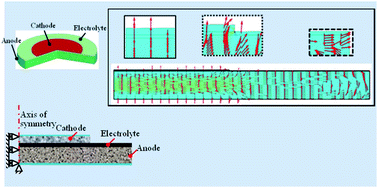
Oxygen ionic transport in conducting ceramics is an important mechanism enabling solid oxide fuel cell (SOFC) technology. The multi-physicochemical processes lead to the fact that the distribution of oxygen vacancy site fraction is not uniform in a positive-electrode electrolyte negative-electrode (PEN) assembly. Different oxygen vacancy concentrations induce different volumetric expansion of ceramics, resulting in complicated chemical–mechanical coupling phenomena and chemical stress in SOFCs. In this research, a mathematical model is developed to study oxygen ionic transport induced chemical stress in an SOFC. The model is validated using experimental polarization curves. Comprehensive simulations are performed to investigate chemical stress distribution in the PEN assembly under different operating conditions and design parameters as well as mechanical constraints. Principal stress analysis is employed to identify the weakest zones in the cell. The Weibull approach is utilized to analyze the failure probability of each component and the elastic energy stored in the cathode layer is employed to evaluate potential delamination failure at the cathode/electrolyte interface. The paper for the first time builds a chemical–mechanical coupling model at a cell level and is an important module complementary to the state-of-the-art electrochemical–thermal–mechanical model of SOFCs.
 Please wait while we load your content...
Please wait while we load your content...

