
Combining block copolymers and hydrogen bonding for poly(lactide) toughening†
Abstract
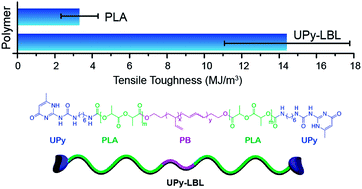
A series of ABA triblock copolymers of poly(D,L-lactide)-b-poly(butadiene)-b-poly(D,L-lactide), LBL, and their 2-ureido-4[1H]-pyrimidinone (UPy) end-functionalized analogue, UPy-LBL, were synthesized. Commercially available hydroxy-terminated poly(butadiene) (HTPB) was used as the macroinitiator for ring-opening polymerizations of D,L-lactide. UPy hydrogen bonding dimerization in solution was confirmed by 1H NMR spectroscopy and by the thermo-reversible properties of this interaction. Mechanical properties of this polymer series revealed that only the incorporation of low molar mass PB did not significantly alter the brittle character of PLA. However, end-functionalization of the triblock copolymers with the UPy moiety effectively increased the strain at break of PLA, producing polymers with average ultimate elongations up to 58%. A relationship between UPy content and mechanical properties was established. The series of polymers with UPy functionalities ranging from 0–2 was tougher than neat PLA; the hemitelechelic polymer was more than four times tougher. To study the physical aging of these systems, uniaxial tensile-tests and DSC experiments were performed. DSC thermograms showed increases in both the glass transition temperature and enthalpy of relaxation as a function of PLA aging time. Tensile experiments demonstrated that UPy-functionalization effectively delayed aging as ductile behavior was only lost after annealing the samples for five days at 40 °C.
 Please wait while we load your content...
Please wait while we load your content...

