
Ce3+ impregnated ZnO: a highly efficient photocatalyst for sunlight mediated mineralization†
Abstract
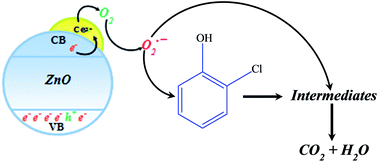
The surface of pre-synthesized hexagonal ZnO was tailored by Ce3+ states. The modified catalyst inveterate enhanced spectral response in the visible region and substantially quenched the luminescence without altering the morphology of the ZnO support. Compared to bare ZnO, the synthesized catalyst exhibited significant high activity both for degradation and mineralization of 2-chlorophenol (2-CP) in sunlight exposure.
 Please wait while we load your content...
Please wait while we load your content...

