
A novel approach to prepare polymer mixed-brushes via single crystal surface patterning†
Abstract
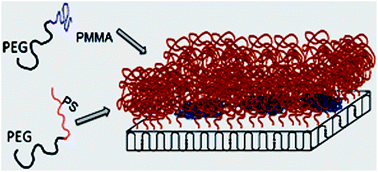
Polymer mixed-brushes with predetermined morphologies as well as with uniform chain length and chain length distribution of various brushes were prepared on a substrate via single crystal surface patterning with a self-seeding procedure from a dilute solution. The required materials including poly(ethylene glycol)-b-polystyrene (PEG-b-PS) and poly(ethylene glycol)-b-poly(methyl methacrylate) (PEG-b-PMMA) with the same crystalline block molecular weight and different amorphous block molecular weights were synthesized via atom transfer radical polymerization (ATRP). Based on various qualities of the employed solvent and various interactions of the substrate with amorphous blocks, the matrix (PS)-dispersed (PMMA) morphologies were obtained on PEG single crystals. The matrix and dispersed phase regions were detectable by atomic force microscopy (AFM) due to differences in height and stiffness. The domain size of dispersed phase decreased with an increase in the molecular weights of PS and PMMA blocks because hindrance against the presence of PEG-b-PMMA chains increased. Despite the domain size of the dispersed phase ranging from 286 to 351 nm, the crystallization temperature did not have a significant effect on the domain size. By increasing the crystallization temperature, the thickness enhancement, and consequently the tethering density enhancement, was more considerable for PS regions, which can be attributed to the lower osmotic pressure of PS chains. Finally, we grew epitaxial structures consisting of PEG-b-PS, PEG-b-PMMA, and homo-PEG layers to verify the determined thicknesses from mixed-brush systems through the conjunction thickness of the corresponding copolymer and homopolymer crystals.
 Please wait while we load your content...
Please wait while we load your content...

