
Syntheses and characterization of new poly(ionic liquid)s designed for CO2 capture
Abstract
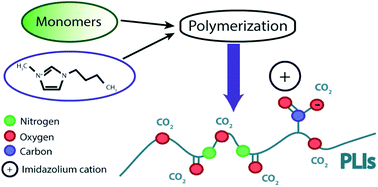
A series of new poly(ionic liquid)s-p(IL)s based on polyurethane structures were synthesized and characterized and their behavior evaluated in CO2 sorption tests under different pressures. The synthesized materials were characterized according to structure, composition, and thermal stability, by techniques such as FTIR, 1H-NMR, TGA and DSC. The CO2 sorption measurements were carried out in a Magnetic Suspension Balance-PTGA and proved that the change of the components of the polymer chain directly indicates the sorption behavior. The best performance for CO2 sorption (75.7 mol% at 20 bar) was achieved with the p(IL) PUA-02a obtained from HDI and PTMG-2000 with [bmim]+ as a counter-ion. The synthesized p(IL)s presenting nitrogenated and polyether structures into the backbone allied to imidazolium counter-cations proved to be worthy of note in the CO2 sorption besides being based on poly(urethane) a versatile and low-cost material. The results also highlighted the good performance of PUA-02 when compared with traditional solvents used in pre-combustion process, as well as the p(IL)s described in the literature.
 Please wait while we load your content...
Please wait while we load your content...

