Synthesis of ionic liquid-supported hypervalent iodine reagent and its application as a ‘catch and release’ reagent for α-substituted acetophenones†
Manoj Kumar Muthyala‡,
Sunita Choudhary‡ and
Anil Kumar*
Department of Chemistry, Birla Institute of Technology and Science, Pilani, Rajasthan 333031, India. E-mail: anilkumar@pilani.bits-pilani.ac.in; Fax: +91-1596-244183; Tel: +91-1596-515652
First published on 4th February 2014
Abstract
A novel imidazolium-based ionic liquid-supported hypervalent iodine reagent has been synthesized and employed for a ‘catch and release’ strategy with substituted acetophenones to generate various α-substituted acetophenones in good to excellent yields. The use of an ionic liquid-supported hypervalent iodine reagent avoids chromatographic separation for the purification of α-substituted acetophenones and thus makes the method greener.
Introduction
High-throughput screening relies on combinatorial chemistry techniques for the rapid production of a library of compounds with adequate purity. It is very challenging to improve the efficiency of the reaction and make the purification process simple and straightforward. Solid-phase organic synthesis (SPOS)1–6 is a widely employed tool in combinatorial synthesis that has facilitated the synthesis of many pharmacologically important compounds and made the purification process more facile. Despite its great success, SPOS is associated with some serious drawbacks such as difficulties in the monitoring of the reaction, the need for extra steps, non-linear kinetics and heterogeneous reaction conditions. This has driven the re-evaluation of supported reagents and scavengers in combinatorial synthesis with the combined advantages of solid- and solution-phase chemistry.7–10 The synthesis of polymer-,11 polyethylene glycol (PEG)-12–14 and silica-supported reagents15–17 is tricky, and it is difficult to determine the amount of reagent that has been grafted on these supports. On the other hand, fluorous-supported reagents are expensive and need specialized solvents to carry out the reactions.18–21In recent years, ionic liquid-supported reagents22–29 have gained considerable interest as promising alternative supported reagents due to their high loading capacity, tunable solubility, homogeneity and easy monitoring of the reaction by various analytical techniques such as NMR, IR and mass spectrometry. Several ionic liquid-supported reagents have been synthesized and used for different organic transformations (Fig. 1), for example, ionic liquid-supported methyl sulfoxide (1) has been used for the Swern oxidation of alcohols,30 ionic liquid-supported tin reagent (2) has been used for Stille cross-coupling,31–33 ionic liquid-supported triphenylphosphine34 (3) has been used as a ligand in C–C coupling reactions, ionic liquid-supported iodobenzene diacetate (IL-IBD)35 (4) has been used for the oxidation of alcohols and sulfides,36 and ionic liquid-supported hydroxyl(tosyl)iodobenzene37 (IL-HTIB) (5) has been used as a tosylating reagent. More recently, we have developed a self-stable and novel ionic liquid-supported sulfonyl azide (6) which has been successfully used as a diazo transfer reagent.38
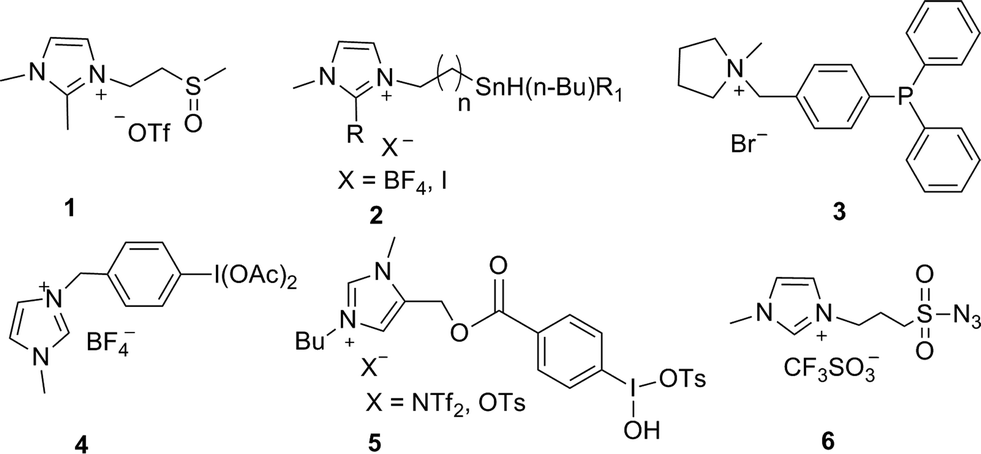 | ||
| Fig. 1 Structure of selected ionic liquid-supported reagents. | ||
Continuing our interest in functionalized ionic liquids,38–42 we envisaged a new synthetic approach where ionic liquid-supported reagents can be employed in a “catch and release (CAR)” strategy. This strategy is a hybrid of scavenging and liquid phase synthesis, where the ionic liquid-supported reagent captures the required substrate and releases the desired product by subsequent reactions. HTIB is an important iodine(III) reagent for the oxytosylation and synthesis of heterocyclic compounds.43–46 In this paper, we wish to report the synthesis of novel ionic liquid-supported hypervalent iodine reagent 11 and demonstrate its application in a “catch and release” strategy for the synthesis of α-substituted acetophenones.
Results and discussion
The synthesis of 11 was achieved from 2,3-dimethylimidazole (7) as shown in Scheme 1. Ionic liquid-supported sulfonic acid (9) was synthesized by the reaction of 7 with sultone (8), followed by acidification with trifluoromethane-sulfonic acid.42 The homogeneous solution of 9 in CH3CN was added to a hot solution of iodobenzene diacetate (10), and the reaction mixture was kept at room temperature for one week to obtain 11. Several attempts were made by varying the reaction conditions to reduce the reaction time to no success, and the best yield of 11 was obtained in a week. The structure of 11 was unambiguously established by spectroscopic data such as 1H, 13C NMR and high-resolution mass spectrometry (HRMS). The 1H NMR spectrum of 11 showed doublets at δ 7.63 and 7.59 for imidazole protons, along with other aromatic and aliphatic protons. In the 13C NMR spectrum, the C1 carbon of the phenyl ring attached to iodine resonated at δ 94.93 and the CF3 carbon of CF3SO3− resonated at δ 121.02 as a quartet, in addition to the other six aliphatic and seven aromatic carbons of the molecule. The molecular ion peak at m/z 453.0345 [M − CF3SO3]+ in the HRMS provided clear evidence towards the structure of 11.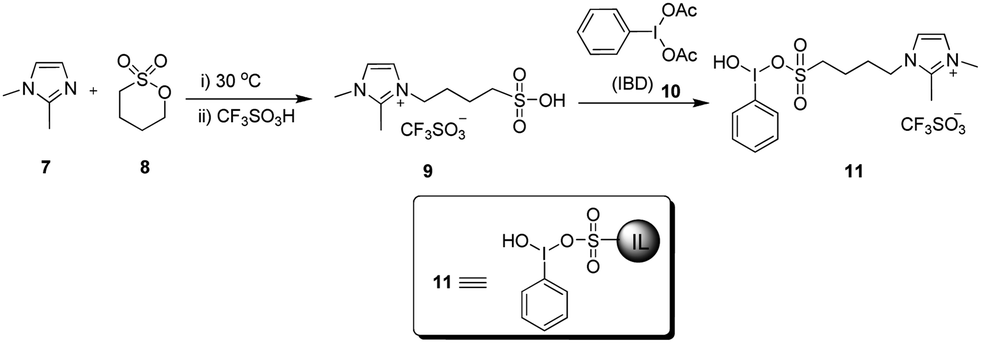 | ||
| Scheme 1 Synthesis of ionic liquid-supported hypervalent iodine. | ||
After successful synthesis of 11, we explored its application as an α-sulfonating agent to capture substituted acetophenones. To optimize the reaction conditions, 4-chloroacetophenone (12a) was selected as a model substrate, and the reaction of 12a with 11 was examined under different reaction conditions as outlined in Table 1. It was found that 12a was completely captured in CH3CN (Table 1, entry 10) using 2 equiv. of 11 at 80 °C after 12 h. The use of other solvents such as THF, CHCl3 and [bmim][BF4] did not result in the efficient capturing of 12a. Although the capturing efficiency was enhanced in solvent-free conditions (Table 1, entry 2 vs 8), the formation of an unidentified impurity was a major drawback under these conditions.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | 11 (mmol) | Temp. (°C) | Time (h) | Conversionb (%) |
| a Reaction conditions: 12a (1 mmol), 11 (mmol as indicated in table), solvent (5 mL), Na2SO4 (0.5 mmol).b Determined based on recovered 12a.c No solvent.d An unidentified impurity was formed along with 13. | |||||
| 1 | —c | 1 | 30 | 12 | 10 |
| 2 | —c | 1 | 80 | 12 | 70d |
| 3 | THF | 1 | Reflux | 18 | Traces |
| 4 | THF | 2 | Reflux | 16 | 22 |
| 5 | [bmim][BF4] | 1 | 80 | 16 | 17 |
| 6 | [bmim][BF4] | 2 | 80 | 12 | 32 |
| 7 | CHCl3 | 2 | Reflux | 14 | 50 |
| 8 | CH3CN | 1 | 80 | 12 | 51 |
| 9 | CH3CN | 2 | 30 | 12 | 27 |
| 10 | CH3CN | 2 | 80 | 12 | 100 |

To confirm the identity of the product 13a, acetonitrile was evaporated and the reaction mixture was run through a short plug of silica using a dichloromethane–methanol mixture (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v) as the eluent. The obtained pure product was analyzed by NMR and mass spectroscopic data. The appearance of a peak at δ 5.70 for the methylene protons attached to the ionic liquid-supported sulfonate group along with the other six aromatic and fourteen aliphatic protons in the 1H NMR spectra, and the peak at δ 191.27 for the carbonyl group, along with the other carbons, in the 13C NMR showed that 12a was ‘captured’ by the ionic liquid. Finally, a peak at m/z 385.098, corresponding to molecular formula, C17H22ClN2O4S+ [M − CF3SO3]+ in the HRMS confirmed the structure of 13a.
:5 v/v) as the eluent. The obtained pure product was analyzed by NMR and mass spectroscopic data. The appearance of a peak at δ 5.70 for the methylene protons attached to the ionic liquid-supported sulfonate group along with the other six aromatic and fourteen aliphatic protons in the 1H NMR spectra, and the peak at δ 191.27 for the carbonyl group, along with the other carbons, in the 13C NMR showed that 12a was ‘captured’ by the ionic liquid. Finally, a peak at m/z 385.098, corresponding to molecular formula, C17H22ClN2O4S+ [M − CF3SO3]+ in the HRMS confirmed the structure of 13a.
To explore the generality of the reaction, acetophenones with both electron-withdrawing and electron-releasing groups were reacted with 11 to yield 13. Most of the substituted acetophenones reacted with 11 smoothly under the given conditions (Table 2), however in the case of 4-methoxy and 4-nitroacetophenones, a small amount of the corresponding benzoic acid (10–12%) was formed along with 13, which was removed by simple washing with diethyl ether.
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | Conversionb (%) | Entry | Ar | Conversionb (%) |
| a Reaction conditions: 12a–h (1.0 mmol), 11 (2.0 mmol), Na2SO4 (0.5 mmol), CH3CN (5.0 mL), 80 °C, 12 h.b Determined based on recovered acetophenone.c The corresponding benzoic acid was formed in 10–12% yield. | |||||
| a | 4-ClC6H4 | 100 | e | 4-CH3OC6H4 | 80c |
| b | 3-ClC6H4 | 100 | f | C10H7 | 100 |
| c | 4-BrC6H4 | 90 | g | 4-NO2C6H4 | 80c |
| d | 4-CH3C6H4 | 90 | h | C4H4S | 100 |

Next, we studied the ‘release’ of acetophenone from 13a using thiocyanate as the anion to give α-thiocyanato-β-ketones (14a). When 13a was treated with KSCN at room temperature for 4 h, the ionic liquid-supported substituted acetophenone was released cleanly with the thiocyanate ion without any side product. Simple extraction with hexane–ethyl acetate (7:3 v/v), followed by washing with water, gave 14a in 92% yield and high purity.
Encouraged with these systematic “catch and release” results for 12a, we focused our attention on developing a simple and straightforward, chromatograpy-free protocol for the synthesis of α-substituted acetophenones (14–16) from 12 (Table 3). Initially, 12a was treated with 11 in dry CH3CN at 80 °C for 12 h in the presence of anhydrous Na2SO4. After completion of the reaction, the reaction mixture was filtered and CH3CN was evaporated under reduced pressure. The residue containing ionic liquid-supported 4-chloroacetophenone (13a) was washed with diethyl ether and subsequently treated with KSCN at room temperature for 4 h. On completion of the reaction, the product was extracted with hexane–ethyl acetate (7:3 v/v), washed with water, dried with anhydrous Na2SO4 and concentrated to give 14a in 92% yield. The purity of 14a was over 98% as indicated by HPLC and NMR (see ESI†).
|
||||
|---|---|---|---|---|
| Entry | Ar | % Yieldb | ||
| 14 | 15 | 16 | ||
| a Reaction conditions: 12 (1.0 mmol), 11 (2.0 mmol), Na2SO4 (0.5 mmol) CH3CN (5 mL), 80 °C, 12 h, followed by Nu (1.0 mmol), 30 °C, 4 h.b Isolated yields. | ||||
| a | 4-ClC6H4 | 92 | 90 | 68 |
| b | 3-ClC6H4 | 70 | 84 | 58 |
| c | 4-BrC6H4 | 80 | 67 | 46 |
| d | 4-CH3C6H4 | 76 | 62 | 52 |
| e | 4-CH3OC6H4 | 65 | 78 | 50 |
| f | C10H7 | 90 | 92 | 68 |
| g | 4-NO2C6H4 | 65 | 70 | 51 |
| h | C4H4S | 75 | 76 | 55 |

On establishing a simple and chromatography-free method for the synthesis of 14a, we utilized different acetophenones (12a–h) with three nucleophiles, –SCN, –N3 and CH3C6H4SO2−, to obtain the corresponding β-substituted acetophenones 14, 15 and 16, respectively (Table 3). As indicated in Table 3, acetophenone with both electron-releasing and electron-withdrawing groups reacted smoothly. α-Thiocyanato-β-ketones (14) and α-azido-β-ketones (15) were obtained in good to excellent yield (62–92%), while β-ketosulfones (16) were obtained in moderate to good yield (46–68%). This may be attributed to their different nucleophilicity. It is worth mentioning that all the compounds were of high purity and no chromatographic separation was performed. The yields of the products (14–16) are based on the acetophenones after two steps. The purification method is simple and involves extraction with hexane–ethyl acetate (7:3 v/v) followed by washing with water and drying with anhydrous sodium sulfate.
Compared to the polymer-bound iodine(III) reagent,47,48 the loading capacity is higher for the ionic liquid-supported reagent, and the reaction can be monitored easily by different spectroscopic techniques. The present method does not require an excess amount of nucleophile to “release” the products as is the case with the polymer-bound reagent, and purification of the products does not require chromatographic separation.
Conclusions
In summary, we have successfully developed an efficient and simple protocol for the synthesis of an ionic liquid-supported hypervalent iodine reagent, and its use has been demonstrated in the generation of α-substituted acetophenones through a “catch and release” strategy. The two-step protocol for the conversion of substituted acetophenones to α-substituted acetophenones involves no chromatographic separation and gives the product in good to excellent yield and high purity. The scope and utility of the developed ionic liquid-supported reagent is general and can be used for the synthesis of various substituted ketones and heterocyclic compounds.Experimental
Synthesis of ionic liquid-supported hypervalent iodine (11)
Ionic liquid-supported sulfonic acid (13 mmol) and IBD (13 mmol) were dissolved in the minimum amount of CH3CN in different conical flasks and heated on a hotplate. The warm ionic liquid-supported sulfonic acid solution was added to the flask containing IBD under hot conditions. The reaction mixture was kept aside for one week. Acetonitrile was removed by a rotary evaporator under reduced pressure, and the resulting mixture was washed with dry DCM (3 × 30 mL) to remove unreacted starting materials to obtain a thick yellow liquid (7.55 g, 96%). 1H NMR (300 MHz, DMSO-d6, CD3OD) δ 8.22 (s, 1H), 7.76 (d, J = 1.1 Hz, 1H), 7.73 (d, J = 1.1 Hz, 1H), 7.62 (d, J = 2.1 Hz, 1H), 7.58 (d, J = 2.0 Hz, 1H), 7.44–7.37 (m, 1H), 7.23–7.15 (m, 2H), 4.14 (t, J = 7.3 Hz, 2H), 3.76 (s, 3H), 2.64 (d, J = 7.5 Hz, 2H), 2.59 (s, 3H), 1.91–1.78 (m, 2H), 1.71–1.58 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 144.6, 137.5, 130.9, 128.0, 123.1, 122.6, 121.2, 121.0 (q, JC–F = 320.25 Hz, OTf), 94.9, 50.7, 34.8, 28.4, 21.9, 9.2; HRMS (ESI-TOF) calculated for C15H22IN2O4S+: 453.0339, found: 453.0356 [M − CF3SO3]+.Synthesis of ionic liquid-supported sulfonates (13a–h)
Acetophenone (1.0 mmol), 11 (2 mmol) and dry Na2SO4 (0.5 mmol) were added to CH3CN (5 mL) and heated at reflux until the acetophenone was completely consumed. The reaction mixture was filtered, CH3CN was removed under reduced pressure and the resulting reaction mixture was purified by silica-gel column chromatography (dichloromethane–methanol 3:1 as the eluent) to obtain the desired compound 13a–h.
13a: Off-white solid (272 mg, 51%); m.p.: 90–96 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.97 (d, J = 8.6 Hz, 2H), 7.70–7.60 (m, 4H), 5.70 (s, 2H), 4.19 (t, J = 6.7 Hz, 2H), 3.75 (s, 3H), 3.55 (t, J = 7.1 Hz, 2H), 2.60 (s, 3H), 1.94–1.70 (m, 4H); 13C NMR (75 MHz, DMSO-d6) δ 191.2, 144.8, 139.5, 132.7, 130.2, 129.5, 122.8, 121.3, 121.1 (q, JC–F = 320.25 Hz, OTf), 71.6, 49.3, 47.2, 35.1, 27.8, 20.3, 9.6; HRMS (ESI-TOF) calculated for C17H22ClN2O4S+: 385.0983, found: 385.0975 [M − CF3SO3]+.
13b: Yellow liquid (265 mg, 50%); 1H NMR (300 MHz, DMSO-d6) δ 8.01–7.97 (m, 1H), 7.92 (d, J = 7.9 Hz, 1H), 7.80 (dd, J = 8.0, 1.1 Hz, 1H), 7.67–7.62 (m, 3H), 5.74 (s, 2H), 4.19 (t, J = 6.9 Hz, 2H), 3.76 (s, 3H), 3.59–3.52 (m, 2H), 2.60 (s, 3H), 1.94–1.76 (m, 4H); 13C NMR (75 MHz, DMSO-d6) δ 189.2, 142.8, 142.6, 133.7, 132.2, 129.3, 126.0, 124.9, 120.7, 119.2, 119.0 (q, JC–F = 320.25 Hz, OTf), 69.6, 47.2, 45.1, 33.0, 25.8, 18.3, 7.5; HRMS (ESI-TOF) calculated for C17H22ClN2O4S+: 385.0983, found: 385.1015 [M − CF3SO3]+.
13c: Pale yellow solid (259 mg, 45%); m.p.: 83–96 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.92–7.86 (m, 2H), 7.83–7.77 (m, 2H), 7.65 (d, J = 2.1 Hz, 1H), 7.62 (d, J = 2.1 Hz, 1H), 5.70 (s, 2H), 4.19 (t, J = 6.8 Hz, 2H), 3.75 (s, 3H), 3.60–3.49 (m, 2H), 2.60 (s, 3H), 1.95–1.72 (m, 4H); 13C NMR (75 MHz, DMSO-d6) δ 191.5, 144.9, 133.1, 132.5, 130.3, 128.7, 122.8, 121.3, 121.1 (q, JC–F = 320.25 Hz, OTf), 71.6, 49.3, 47.2, 35.1, 27.8, 20.4, 9.6; HRMS (ESI-TOF) calculated for C17H22BrN2O4S+: 429.0478, found: 429.0512 [M − CF3SO3]+ and 431.0489 [M + 2 − CF3SO3]+.
13d: Yellow solid (231 mg, 45%); m.p.: 64–70 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.86 (d, J = 8.2 Hz, 2H), 7.64 (dd, J = 8.9, 2.1 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 5.68 (s, 2H), 4.19 (t, J = 6.8 Hz, 2H), 3.76 (s, 3H), 3.63–3.48 (m, 2H), 2.60 (s, 3H), 2.40 (s, 3H), 1.96–1.78 (m, 4H); 13C NMR (75 MHz, DMSO-d6) δ 191.6, 145.3, 144.9, 131.5, 129.9, 128.4, 122.8, 121.3, 121.1 (q, JC–F = 320.25 Hz, OTf), 71.6, 49.3, 47.2, 35.1, 27.8, 21.7, 20.4, 9.6; HRMS (ESI-TOF) calculated for C18H25N2O4S+: 365.1530, found: 365.1555 [M − CF3SO3]+.
13e: Thick light brown liquid (211 mg, 40%); 1H NMR (300 MHz, DMSO-d6) δ 7.94 (d, J = 9.0 Hz, 2H), 7.63 (dd, J = 8.9, 2.1 Hz, 2H), 7.12–7.06 (m, 2H), 5.64 (s, 2H), 4.21–4.11 (m, 2H), 3.86 (s, 3H), 3.75 (s, 3H), 3.56–3.47 (m, 2H), 2.59 (s, 3H), 1.95–1.78 (m, 4H); 13C NMR (75 MHz, DMSO-d6) δ 190.4, 164.3, 144.8, 130.7, 126.8, 122.8, 121.3, 121.1 (q, JC–F = 320.25 Hz, OTf), 114.6, 71.4, 56.1, 49.4, 47.2, 35.1, 27.8, 20.4, 9.5; HRMS (ESI-TOF) calculated for C18H25N2O5S+: 381.1479, found: 381.1504 [M − CF3SO3]+.
13f: Brown liquid (373 mg, 68%); 1H NMR (300 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.16–7.95 (m, 4H), 7.76–7.62 (m, 4H), 5.88 (s, 2H), 4.21 (t, J = 6.7 Hz, 2H), 3.77 (s, 3H), 3.60 (t, J = 7.1 Hz, 2H), 2.61 (s, 3H), 2.02–1.70 (m, 4H); 13C NMR (75 MHz, DMSO-d6) δ 192.0, 144.9, 135.9, 132.4, 131.3, 130.6, 130.0, 129.6, 129.1, 128.3, 127.7, 123.5, 122.8, 121.3, 121.3 (q, JC–F = 320.25 Hz, OTf), 71.7, 49.3, 47.2, 35.1, 27.9, 20.4, 9.6; HRMS (ESI-TOF) calculated for C21H25N2O4S+: 401.1530, found: 401.1524 [M − CF3SO3]+.
13g: Brown solid (218 mg, 40%); m.p.: 85–90 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.39 (d, J = 8.7 Hz, 2H), 8.19 (d, J = 8.8 Hz, 2H), 7.64 (dd, J = 7.5, 1.8 Hz, 2H), 5.78 (s, 2H), 4.19 (t, J = 6.7 Hz, 2H), 3.76 (s, 3H), 3.56 (t, J = 7.0 Hz, 2H), 2.60 (s, 3H), 1.98–1.73 (m, 4H); 13C NMR (75 MHz, DMSO-d6) δ 191.5, 150.8, 144.9, 138.7, 129.9, 124.4, 122.8, 121.3, 121.2 (q, JC–F = 320.25 Hz, OTf), 71.9, 49.3, 47.2, 35.1, 27.8, 20.3, 9.6; HRMS (ESI-TOF) calculated for C17H21N3O6S+: 395.1240, found: 395.1252 [M − CF3SO3]+.
13h: Thick light brown liquid (252 mg, 50%); 1H NMR (300 MHz, DMSO-d6) δ 8.14 (dd, J = 4.9, 0.7 Hz, 1H), 8.08–8.01 (m, 1H), 7.64 (dd, J = 8.2, 2.0 Hz, 2H), 7.32 (dd, J = 4.7, 4.0 Hz, 1H), 5.60 (s, 2H), 4.19 (t, J = 6.7 Hz, 2H), 3.75 (s, 3H), 3.60–3.47 (m, 2H), 2.59 (s, 3H), 1.95–1.71 (m, 4H); 13C NMR (75 MHz, DMSO-d6) δ 185.4, 144.9, 140.0, 136.5, 134.5, 129.5, 122.8, 121.3, 121.1 (q, JC–F = 320.25 Hz, OTf), 70.8, 49.4, 47.2, 35.1, 27.8, 20.4, 9.6; HRMS (ESI-TOF) calculated for C15H21N2O4S2+: 357.0937, found: 357.0949 [M − CF3SO3]+.
General procedure for the synthesis of α-substituted acetophenones (14–16)
To a solution of substituted acetophenone (12) (1.0 mmol) and 11 (2.0 mmol) in CH3CN, anhydrous Na2SO4 (0.5 mmol) was added and the resulting mixture was heated until the acetophenone was completely consumed (monitored by TLC). After completion of the reaction, the reaction mixture was filtered and CH3CN was evaporated under reduced pressure. The residue was washed with ether (3 × 10 mL), and the viscous ionic liquid-supported sulfonate obtained (13) was treated with KSCN/NaN3/CH3C6H4SO2Na (1.0 mmol) and stirred vigorously at room temperature under solvent-free conditions. After completion of the reaction, the product was extracted with hexane–ethyl acetate (3 × 10 mL, 7:3 v/v), leaving behind the ionic liquid 9 which is insoluble in this solvent mixture. Thus, the product was extracted in hexane–ethyl acetate and washed with water. The organic layers were dried over anhydrous sodium sulfate and concentrated to obtain the pure product (14–16) without chromatographic purification.
Acknowledgements
The authors sincerely thank the Council of Scientific and Industrial Research (CSIR), New Delhi for research funding [Grant No. 02(0115)/13/EMR-II]. MMK and SC thank CSIR for providing the research fellowship.Notes and references
- P. H. Seeberger and W.-C. Haase, Chem. Rev., 2000, 100, 4349–4394 CrossRef CAS PubMed.
- U. Boas, J. Brask and K. J. Jensen, Chem. Rev., 2009, 109, 2092–2118 CrossRef CAS PubMed.
- F. Guillier, D. Orain and M. Bradley, Chem. Rev., 2000, 100, 2091–2158 CrossRef CAS PubMed.
- R. E. Sammelson and M. J. Kurth, Chem. Rev., 2000, 101, 137–202 CrossRef PubMed.
- S. Booth, P. H. H. Hermkens, H. C. J. Ottenheijm and D. C. Rees, Tetrahedron, 1998, 54, 15385–15443 CrossRef CAS.
- L. A. Sloan and D. J. Procter, Chem. Soc. Rev., 2006, 35, 1221–1229 RSC.
- R. S. Varma, Green Chem., 1999, 1, 43–55 RSC.
- S. Kobayashi, Chem. Soc. Rev., 1999, 28, 1–15 RSC.
- J. Lu and P. H. Toy, Chem. Rev., 2009, 109, 815–838 CrossRef CAS PubMed.
- T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325–3344 CrossRef CAS PubMed.
- S. V. Ley, A. G. Leach and R. I. Storer, J. Chem. Soc., Perkin Trans. 1, 2001, 358–361 RSC.
- D. J. Gravert and K. D. Janda, Chem. Rev., 1997, 97, 489–510 CrossRef CAS PubMed.
- P. H. Toy and K. D. Janda, Acc. Chem. Res., 2000, 33, 546–554 CrossRef CAS PubMed.
- A. D. Wentworth, P. Wentworth, U. F. Mansoor and K. D. Janda, Org. Lett., 2000, 2, 477–480 CrossRef CAS PubMed.
- T. Fey, H. Fischer, S. Bachmann, K. Albert and C. Bolm, J. Org. Chem., 2001, 66, 8154–8159 CrossRef CAS PubMed.
- A. Michaud, G. Gingras, M. Morin, F. Béland, R. Ciriminna, D. Avnir and M. Pagliaro, Org. Process Res. Dev., 2007, 11, 766–768 CrossRef CAS.
- R. S. Varma, Tetrahedron, 2002, 58, 1235–1255 CrossRef CAS.
- T. Miura, K. Goto, D. Hosaka and T. Inazu, Angew. Chem., Int. Ed., 2003, 42, 2047–2051 CrossRef CAS PubMed.
- S. Luo, L. Zhu, A. Talukdar, G. Zhang, X. Mi, J.-P. Cheng and P. G. Wang, Mini-Rev. Org. Chem., 2005, 2, 177–202 CrossRef CAS.
- A. P. Dobbs and M. R. Kimberley, J. Fluorine Chem., 2002, 118, 3–17 CrossRef CAS.
- C. Zong, A. Venot, O. Dhamale and G.-J. Boons, Org. Lett., 2013, 15, 342–345 CrossRef CAS PubMed.
- M. A. Zolfigol, A. Khazaei, A. R. Moosavi-Zare, A. Zare, H. G. Kruger, Z. Asgari, V. Khakyzadeh and M. Kazem-Rostami, J. Org. Chem., 2012, 77, 3640–3645 CrossRef CAS PubMed.
- F. D'Anna, S. Marullo and R. Noto, J. Org. Chem., 2008, 73, 6224–6228 CrossRef CAS PubMed.
- A. Kamal and G. Chouhan, Tetrahedron Lett., 2005, 46, 1489–1491 CrossRef CAS PubMed.
- L. D. S. Yadav, R. Patel, V. K. Rai and V. P. Srivastava, Tetrahedron Lett., 2007, 48, 7793–7795 CrossRef CAS PubMed.
- C. Zhu, A. Yoshimura, Y. Wei, V. N. Nemykin and V. V. Zhdankin, Tetrahedron Lett., 2012, 53, 1438–1444 CrossRef CAS PubMed.
- W. Qian, E. Jin, W. Bao and Y. Zhang, Angew. Chem., Int. Ed., 2005, 44, 952–955 CrossRef CAS PubMed.
- Y. Suzuki, M. Iinuma, K. Moriyama and H. Togo, Synlett, 2012, 1250–1256 CAS.
- M. Iinuma, K. Moriyama and H. Togo, Tetrahedron, 2013, 69, 2961–2970 CrossRef CAS PubMed.
- X. He and T. H. Chan, Tetrahedron, 2006, 62, 3389–3394 CrossRef CAS PubMed.
- J. Vitz, D. H. Mac and S. Legoupy, Green Chem., 2007, 9, 431–433 RSC.
- N. Louaisil, P. D. Pham, F. Boeda, D. Faye, A.-S. Castanet and S. Legoupy, Eur. J. Org. Chem., 2011, 2011, 143–149 CrossRef.
- P. Dien Pham, J. Vitz, C. Chamignon, A. Martel and S. Legoupy, Eur. J. Org. Chem., 2009, 3249–3257 CrossRef.
- Y. Imura, N. Shimojuh, Y. Kawano and H. Togo, Tetrahedron, 2010, 66, 3421–3426 CrossRef CAS PubMed.
- C. Fang, W. Qian and W. Bao, Synlett, 2008, 2529–2531 CAS.
- W. Qian and L. Pei, Synlett, 2006, 0709–0712 CrossRef CAS PubMed.
- S. T. Handy and M. Okello, J. Org. Chem., 2005, 70, 2874–2877 CrossRef CAS PubMed.
- M. K. Muthyala, S. Choudhary and A. Kumar, J. Org. Chem., 2012, 77, 8787–8791 CrossRef CAS PubMed.
- M. K. Muthayala, B. S. Chhikara, K. Parang and A. Kumar, ACS Comb. Sci., 2011, 14, 60–65 CrossRef PubMed.
- M. K. Muthayala and A. Kumar, ACS Comb. Sci., 2011, 14, 5–9 CrossRef PubMed.
- M. K. Muthyala, B. S. Chhikara, K. Parang and A. Kumar, Can. J. Chem., 2012, 90, 290–297 CrossRef CAS.
- A. Kumar, M. K. Muthyala, S. Choudhary, R. K. Tiwari and K. Parang, J. Org. Chem., 2012, 77, 9391–9396 CrossRef CAS PubMed.
- J.-M. Chen and X. Huang, Synthesis, 2004, 1577–1580 CAS.
- R. M. Moriarty, R. Penmasta, A. K. Awasthi, W. R. Epa and I. Prakash, J. Org. Chem., 1989, 54, 1101–1104 CrossRef CAS.
- J. S. Lodaya and G. F. Koser, J. Org. Chem., 1988, 53, 210–212 CrossRef CAS.
- R. Aggarwal and G. Sumran, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2006, 45, 2690–2695 Search PubMed.
- K. C. Nicolaou, P. S. Baran and Y.-L. Zhong, J. Am. Chem. Soc., 2000, 122, 10246–10248 CrossRef CAS.
- K. C. Nicolaou, T. Montagnon, T. Ulven, P. S. Baran, Y. L. Zhong and F. Sarabia, J. Am. Chem. Soc., 2002, 124, 5718–5728 CrossRef CAS PubMed.
- Z. Guo, S. Liu, Y. Yang, T. Wang and L. Wu, Bull. Korean Chem. Soc., 2011, 32, 3760–3763 CrossRef.
- C. C. Palsuledesai, S. Murru, S. K. Sahoo and B. K. Patel, Org. Lett., 2009, 11, 3382–3385 CrossRef CAS PubMed.
- J. S. Yadav, B. V. Subba Reddy, U. V. Subba Reddy and A. D. Krishna, Tetrahedron Lett., 2007, 48, 5243–5246 CrossRef CAS PubMed.
- O. Prakash, H. Kaur, H. Batra, N. Rani, S. P. Singh and R. M. Moriarty, J. Org. Chem., 2001, 66, 2019–2023 CrossRef CAS PubMed.
- O. Prakash, K. Pannu, R. Prakash and A. Batra, Molecules, 2006, 11, 523–527 CrossRef CAS PubMed.
- H. Takeuchi, S. Yanagida, T. Ozaki, S. Hagiwara and S. Eguchi, J. Org. Chem., 1989, 54, 431–434 CrossRef CAS.
- J. H. Boyer and D. Straw, J. Am. Chem. Soc., 1952, 74, 4506–4508 CrossRef CAS.
- R. Moumne, V. Larue, B. Seijo, T. Lecourt, L. Micouin and C. Tisne, Org. Biomol. Chem., 2010, 8, 1154–1159 CAS.
- D. Kumar, S. Sundaree, V. S. Rao and R. S. Varma, Tetrahedron Lett., 2006, 47, 4197–4199 CrossRef CAS PubMed.
- H. Qian and X. Huang, Synthesis, 2006, 1934–1936 CAS.
- V. Nair, A. Augustine, S. Panicker, T. D. Suja and S. Mathai, Res. Chem. Intermed., 2003, 29, 213–226 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: copies of 1H and 13C NMR for compounds 11, 13 and 14–16, and HPLC analysis of 14a. See DOI: 10.1039/c4ra00063c |
| ‡ Both authors contributed equally |
| This journal is © The Royal Society of Chemistry 2014 |