 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLewis acid promoted C–C and copper-catalyzed C–O bond formation: synthesis of neoflavans†
B.
Suchand
,
J.
Krishna
,
K.
Mritunjoy
and
G.
Satyanarayana
*
Department of Chemistry, Indian Institute of Technology (IIT) Hyderabad, Ordnance Factory Estate Campus, Yeddumailaram - 502 205, Medak District, Andhra Pradesh, India. E-mail: gvsatya@iith.ac.in; Fax: +91 40 2301 6032; Tel: +91 40 2301 6054
First published on 31st January 2014
Abstract
An intramolecular [Cu]-catalyzed C–O bond formation for the synthesis of neoflavans is presented. Lewis acid promoted Friedel–Crafts Michael addition of electron rich aromatic systems onto the double bond of the cinnamate ester was employed to furnish a β-diaryl ester. Electrophilic aromatic bromination of the β-diaryl ester and reduction/Grignard addition furnished the required precursor alcohols. The method is applicable to the synthesis of neoflavans containing tertiary as well as quaternary carbon centers. Significantly, the neoflavan substructures are present in biologically active compounds.
Introduction
The neoflavans (4-aryl-3,4-dihydro-2H-chromenes) or substituted chromans are ubiquitous substructures present in biologically active natural compounds.1–3 For example, the Dalbergia plant species are used in traditional Chinese medicine for curing inflammation, blood disorders and ischemia.4 Similarly, the simple neoflavene dalbergichromene, isolated from the stembark of Dalbergia species,5,6 shows medicinal activity. Also, the chroman natural product centchroman exhibits antifertility properties as an estrogen antagonist, which was isolated from the methanolic extracts of A. indicum.7 In addition, 3-hydroxy-4-arylchroman is also observed as a substructure in 4-arylflavan-3-ol, isolated from the South African plant Nelia meyeri.8 Further, the flavonoid myristinin A possessing the 4-arylchroman part structure is a potent polymerase beta inhibitor (Fig. 1).9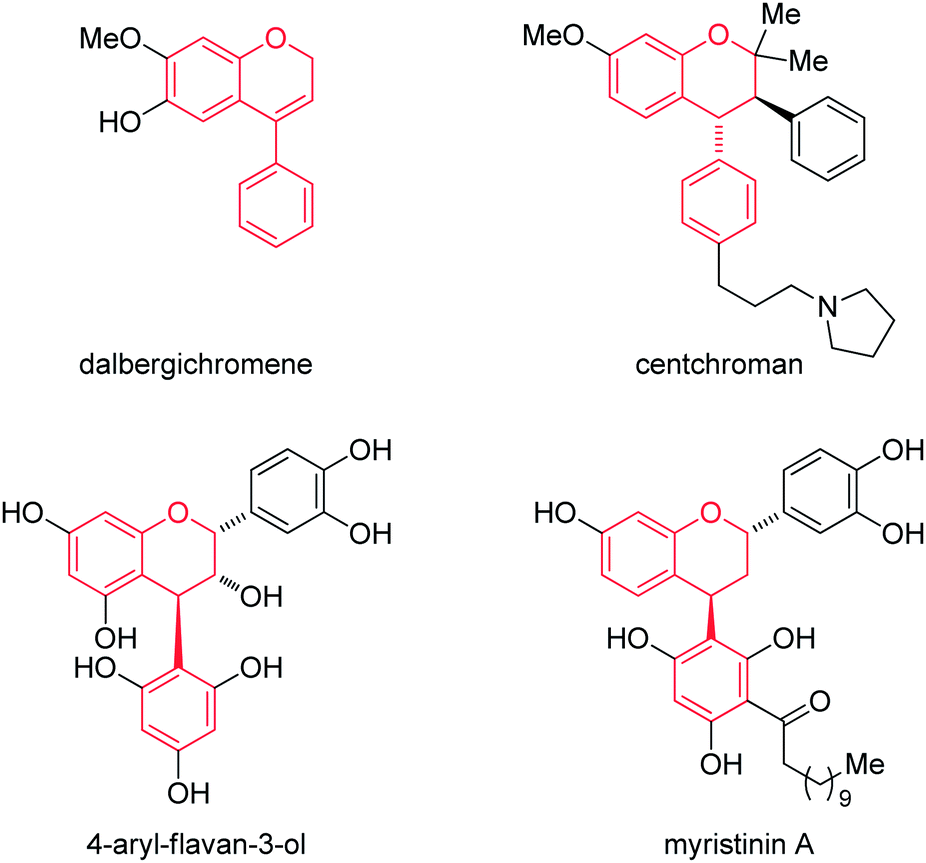 | ||
| Fig. 1 Representative examples of naturally occurring neoflavan natural products containing neoflavan scaffolds. | ||
Owing to the interesting structural features and important biological properties of neoflavans/flavenes, there are a number of reports towards their synthesis.10 Notably, Wang et al.reported enantioselective synthesis of chromans and dihydrobenzopyrans by employing Friedel–Crafts alkylation and cyclization of 1-naphthol and α,β-unsaturated aldehydes.10c Benzopyrans were produced by Pettus and co-workers using enantioselective [4 + 2] cycloaddition between ortho-quinone methides (o-QM) and enol ethers.10f The research group of Sames et al. developed new method for the construction of the chromene core by means of Pt(IV)-catalyzed intramolecular hydroarylation of arene–alkyne substrates.10g The research group of Tunge disclosed a three-step stereoselective reduction as the key step for the synthesis of diarylchromans from phenols and cinnamic acid.10h To the best of our knowledge, thus far there is no report on the synthesis of neoflavans possessing a quaternary C-4 carbon atom.
In continuation of our research interest in transition-metal catalysis,11 recently we have disclosed an efficient three-step strategy for the synthesis of flavans and benzoxepines by employing [Pd]-catalyzed C–C and [Cu]-catalyzed C–O bond-forming reactions as the key steps.12 Herein, we present a practical method for the synthesis of neoflavans, wherein Lewis acid (FeCl3) promoted Friedel–Crafts Michael addition and an intramolecular [Cu]-catalyzed Buchwald–Hartwig coupling reaction are the key transformations involved in the strategy.
Results and discussion
We envisioned that the substituted neoflavans 5 and 7 can be obtained by a key [Cu]-mediated intramolecular Buchwald–Hartwig C–O bond formation between aryl bromide and hydroxy (primary/tertiary) functionality of 4 and 6 respectively. The required primary/tertiary alcohols 4 and 6 were produced from the corresponding esters, which were in turn accessed by employing Lewis acid (FeCl3) induced controlled Friedel–Crafts alkylation (Michael addition type) of an electron rich external arene on ethyl cinnamates 1 followed by preferential electrophilic bromination of the electron rich aromatic ring particularly at the ortho-position to the ester tether (Scheme 1). It is worth mentioning that the controlled Friedel–Crafts alkylation was observed, particularly, by the Lewis acid (FeCl3) in our previous study, which formed the basic foundation for this work.13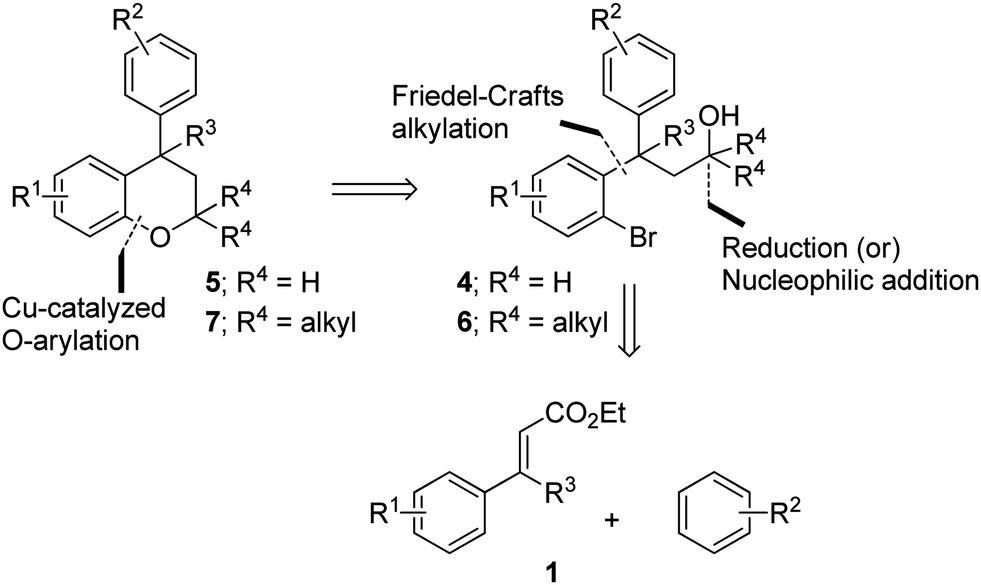 | ||
| Scheme 1 Retrosynthetic plan for flavans 5 and 7 from cinnamates 1. | ||
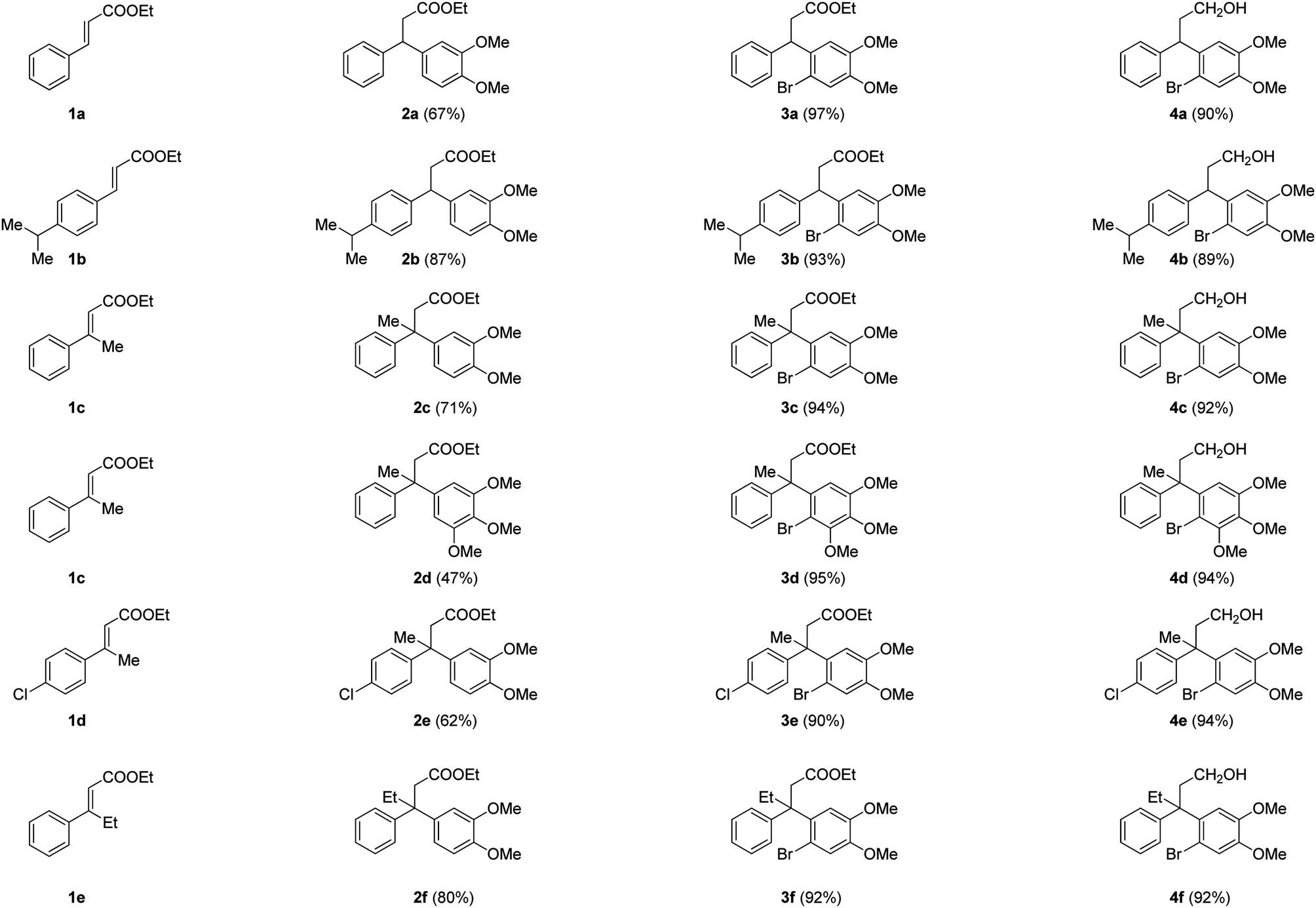
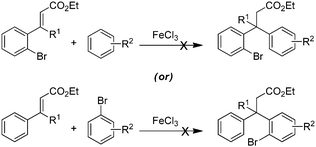
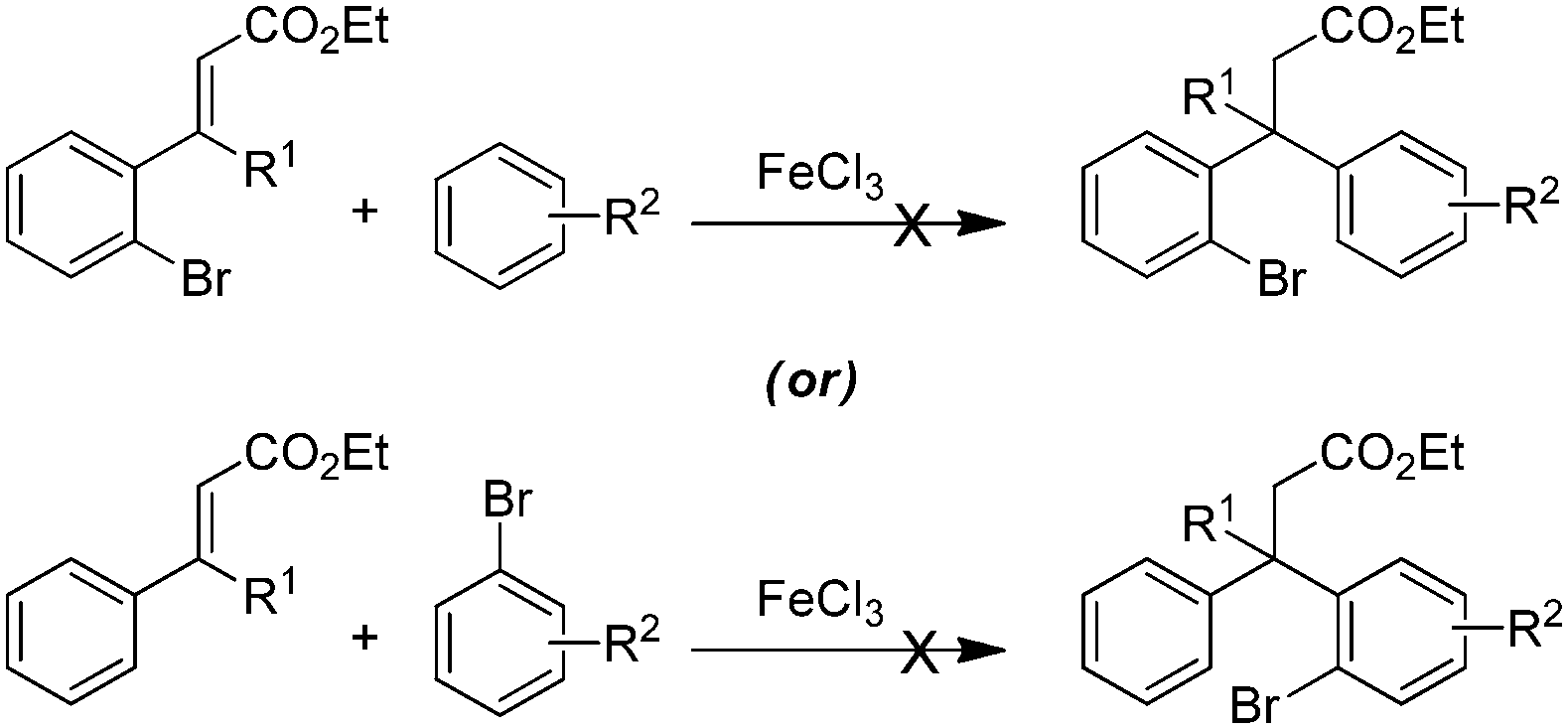
The synthetic study began with the controlled Friedel–Crafts alkylation (i.e. the reaction impeded after Friedel–Crafts Michael addition without allowing it to proceed to subsequent intramolecular acylation) of ethyl cinnamates 1 with electron rich aromatic systems such as 1,2-dimethoxybenzene or 1,2,3-trimethoxybenzene. Thus, treatment of simple as well as β-alkyl substituted ethyl cinnamates 1 with the external arene in the presence of Lewis acid (FeCl3, 3 equiv.), at ambient temperature, furnished the β-arylated esters 2a–2f, containing tertiary/quaternary carbon atom, in fair to very good yields (Table 1).13 It is worth mentioning that the use of DCE as a medium was found to be more general than CH2Cl2 in order to give good yields of the products 2. However, the same Friedel–Crafts Michael addition either with external arene on 2-bromocinnamate or with external bromoarene on simple cinnamate failed to furnish the corresponding product.14 Preferential bromination of the electron rich aromatic ring under the standard electrophilic bromination conditions, provided the esters 3a–3f in excellent yields as shown in Table 1. It is worth mentioning that when both the aromatic rings were electron rich, the bromination was not selective and gave a mixture of brominated products. Next, reduction of the ester function was required to provide the corresponding alcohols for the planned metal mediated cyclization. The esters 3a–3f were reduced with LiAlH4, and the precursor primary alcohols 4a–4f yielded in near quantitative yields, in a controlled manner without affecting the bromo-substituent (Table 1).
|
|
|---|
| a Isolated yields of chromatographically pure products. |
|
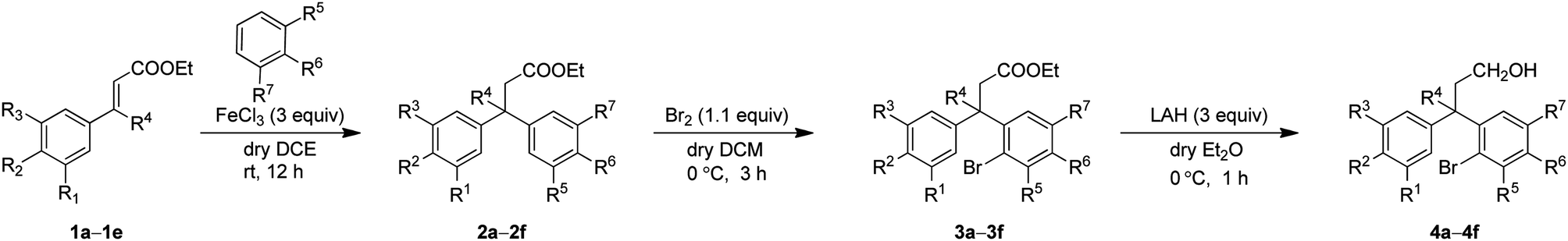
With the precursor alcohols 4 in hand, the key transition metal catalyzed intramolecular C–O bond formation was explored. In this regard, we employed the same reaction conditions [CuI (20 mol%)/2,2-bipyridyl (20 mol%), base KOtBu (3 equiv.) in hot DMF (120 °C) for 24 h] which were successful in our previous reports for the synthesis of flavans.12 As expected, the reaction was quite successful and furnished the neoflavan 5a in very good yield.
Since the above [Cu]-catalyzed C–O bond forming reaction conditions were successful for the construction of neoflavan 5a (Table 2), these conditions were applied to the other alcohols 4b–4f as well. Agreeably, these conditions were found to be versatile and gave the cyclized neoflavans 5b–5f (Table 2) possessing tertiary and quaternary C-4 carbon centers, in very good yields.
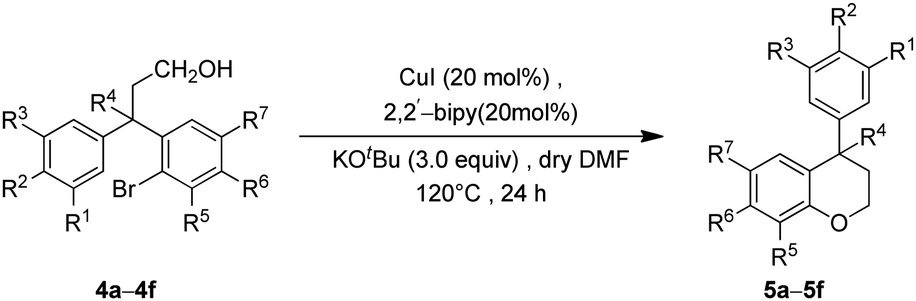
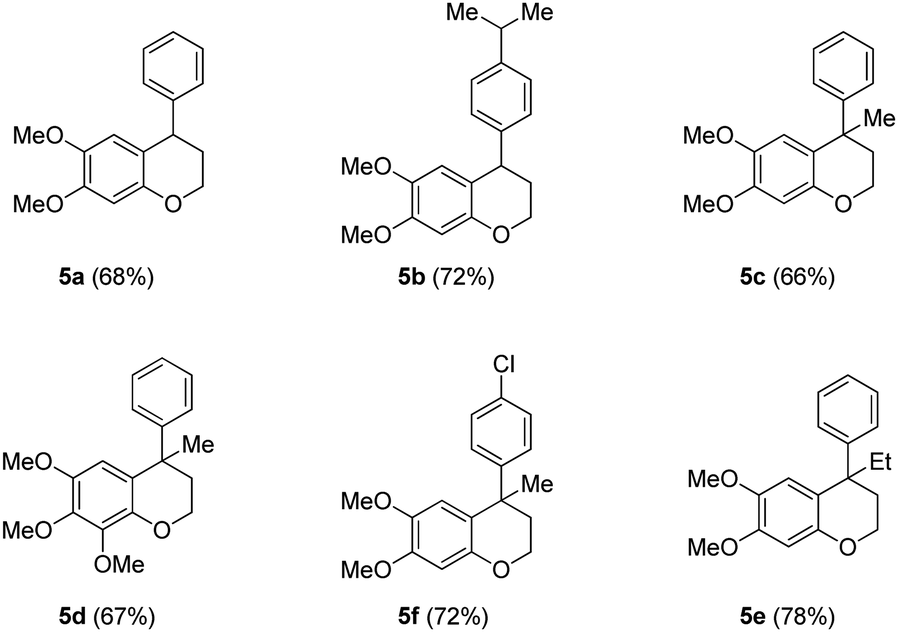
After successful synthesis of various neoflavan derivatives 5a–5f with varied functionalities on either aromatic ring starting from primary alcohol precursors 4a–4f, to check the scope and limitations of the method, we turned our attention to the synthesis of tertiary alcohols. Thus, the reaction of β-diaryl esters 3 with Grignard reagents furnished the corresponding tertiary alcohols 6 as described in Table 3.
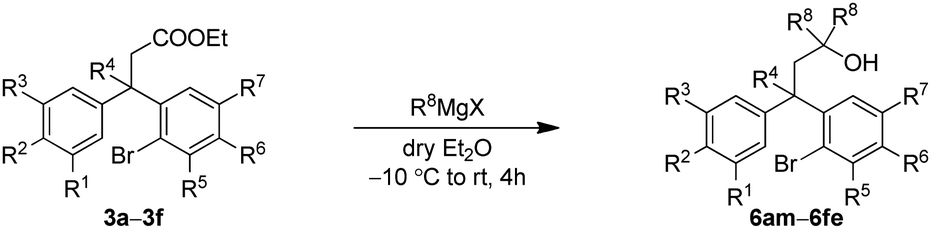
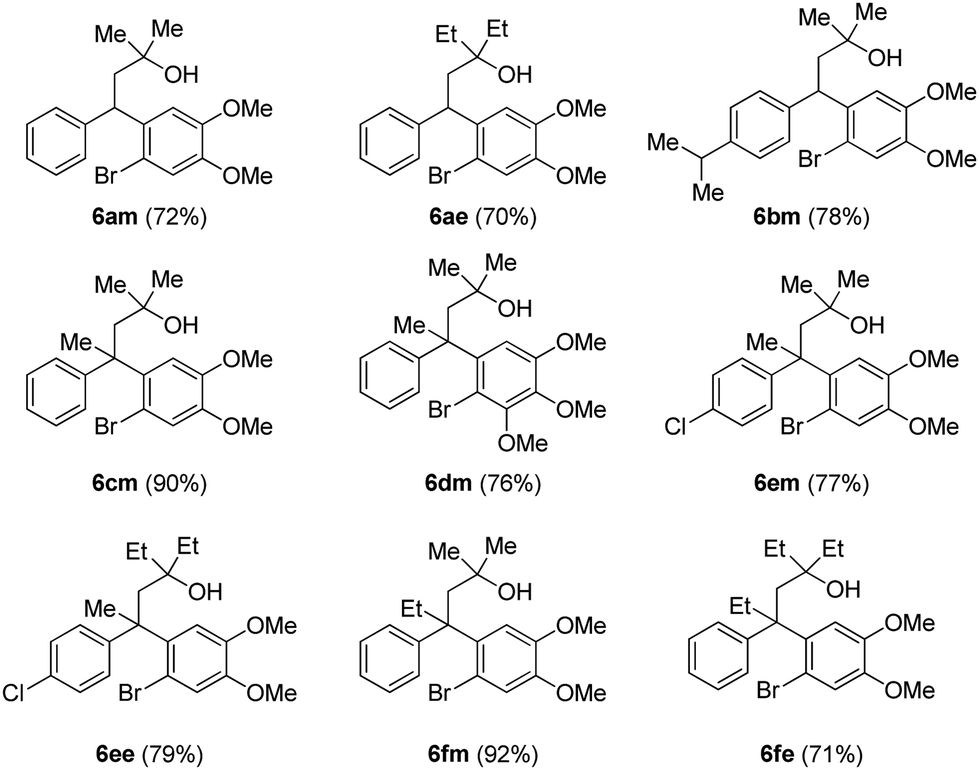
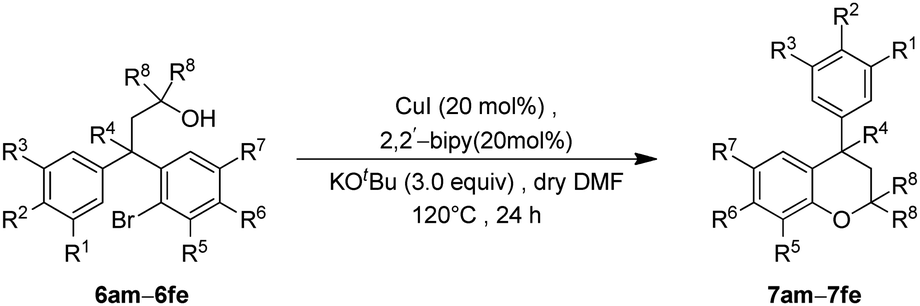
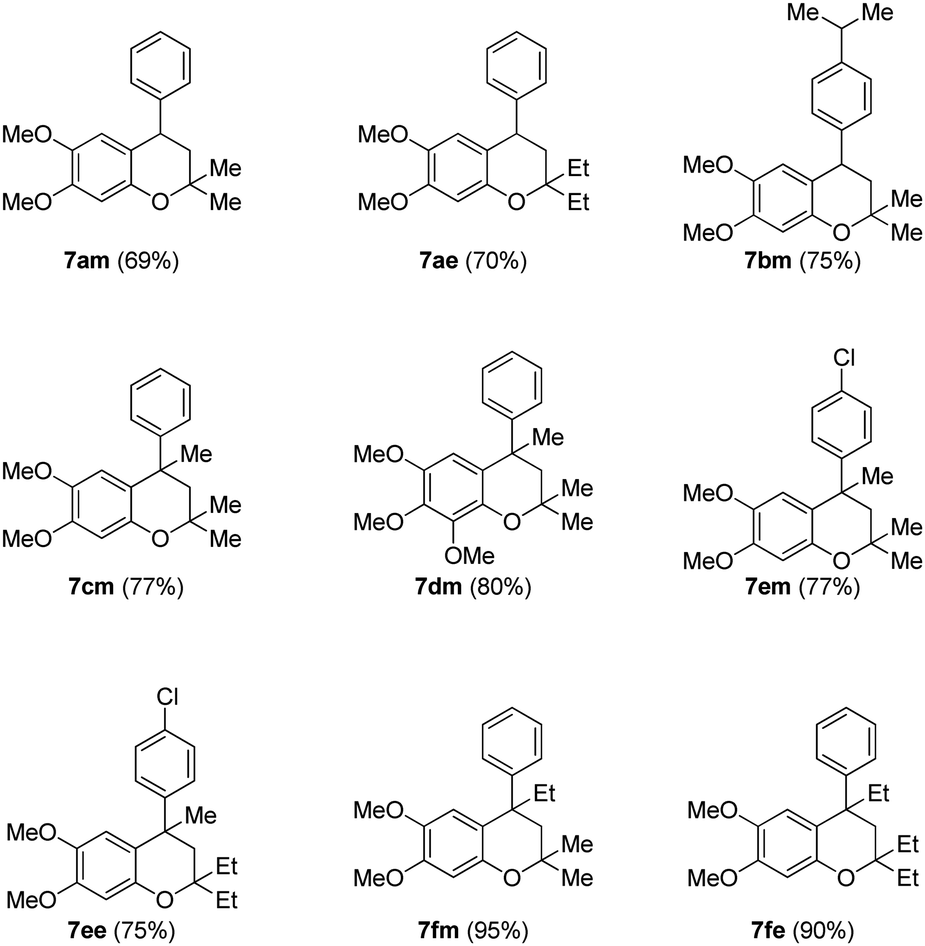
Finally, the key intramolecular C–O bond formation catalyzed by a copper metal complex was explored. As expected, the method was found to be quite successful on the tertiary alcohols as well and generated the cyclized neoflavans, 7am–7fe, in very good yields (Table 4).
Conclusions
In summary, we have developed a simple and practically applicable four-step strategy for the synthesis of functionalized neoflavans. Intermolecular Friedel–Crafts alkylation (C–C bond formation) and intramolecular [Cu]-catalyzed C–O bond formation were applied as key steps of the strategy. The strategy is efficient and amenable to the synthesis of neoflavans containing tertiary as well quaternary carbon centers.Acknowledgements
Financial support by the Department of Science and Technology [(DST), CHE/2010-11/006/DST/GSN], New Delhi is gratefully acknowledged. J. K. thanks CSIR, New Delhi, for the award of a research fellowship.Notes and references
- W. B. Eyton, W. D. Ollis, I. O. Sutherland, O. R. Gottlieb, M. T. Magalhaes and L. M. Jackman, Tetrahedron, 1965, 21, 2683 CrossRef CAS.
- D. M. X. Donnelly and G. Boland, The Flavonoids: Advances in Research since 1986, ed. J. B. Harborne, Chapman & Hall, London, 1994, p. 239 Search PubMed.
- (a) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed; (b) H. C. Shen, Tetrahedron, 2009, 65, 3931 CrossRef CAS PubMed.
- S. C. Chan, Y. S. Chang and S. C. Kuo, Phytochemistry, 1997, 46, 947 CrossRef CAS.
- S. K. Mukerjee, T. Saroja and T. R. Seshadri, Tetrahedron, 1971, 27, 799 CrossRef CAS.
- V. K. Dhingra, S. K. Mukerjee, T. Saroja and T. R. Seshadri, Phytochemistry, 1971, 10, 2551 CrossRef CAS.
- M. S. Sankaran and M. R. N. Prasad, Contraception, 1974, 9, 279 CrossRef CAS.
- H. Kolodziej, Phytochemistry, 1984, 23, 1745 CrossRef CAS.
- (a) J.-Z. Deng, S. R. Starck, S. Li and S. M. Hecht, J. Nat. Prod., 2005, 68, 1625 CrossRef CAS PubMed; (b) D. J. Maloney, J.-Z. Deng, S. R. Starck, Z. Gao and S. M. Hecht, J. Am. Chem. Soc., 2005, 127, 4140 CrossRef CAS PubMed; (c) S. Sawadjoon, P. Kittakoop, K. Kirtikara, V. Vichai and M. Y. Tanticharoen, J. Org. Chem., 2002, 67, 5470 CrossRef CAS PubMed.
- (a) D. M. X. Donnelly, J.-P. Finet, P. J. Guiry and K. Nesbitt, Tetrahedron, 2001, 57, 413 CrossRef CAS; (b) S.-R. Li, L.-Y. Chen, J.-C. Tsai, J.-Y. Tzeng, I.-L. Tsai and E.-C. Wang, Tetrahedron Lett., 2007, 48, 2139 CrossRef CAS PubMed; (c) L. Hong, L. Wang, W. Sun, K. Wong and R. Wang, J. Org. Chem., 2009, 74, 6881 CrossRef CAS PubMed; (d) J. Barluenga, M. Trincado, E. Rubio and J. M. González, J. Am. Chem. Soc., 2004, 126, 3416 CrossRef CAS PubMed; (e) W. Quaglia, M. Pigini, A. Piergentili, M. Giannella, F. Gentili, G. Marucci, A. Carrieri, A. Carotti, E. Poggesi, A. Leonardi and C. Melchiorre, J. Med. Chem., 2002, 45, 1633 CrossRef CAS PubMed; (f) C. Selenski and T. R. R. Pettus, J. Org. Chem., 2004, 69, 9196 CrossRef CAS PubMed; (g) S. J. Pastine, S. W. Youn and D. Sames, Org. Lett., 2003, 5, 1055 CrossRef CAS PubMed; (h) K. Li, K. Vanka, W. H. Thompson and J. A. Tunge, Org. Lett., 2006, 8, 4711 CrossRef CAS PubMed; (i) B. D. Gallagher, B. R. Taft and B. H. Lipshutz, Org. Lett., 2009, 11, 5374 CrossRef CAS PubMed; (j) J. D. Chambers, J. Crawford, H. W. R. Williams, C. Dufresne, J. Scheigetz, M. A. Bernstein and C. K. Lau, Can. J. Chem., 1992, 70, 1717 CrossRef CAS; (k) G. Liu and X. Lu, Tetrahedron, 2008, 64, 7324 CrossRef CAS PubMed; (l) A. Volonterio and M. Zanda, Tetrahedron Lett., 2005, 46, 8723 CrossRef CAS PubMed.
- (a) A. G. K. Reddy, J. Krishna and G. Satyanarayana, Synlett, 2011, 1756 CAS; (b) J. Krishna, A. G. K. Reddy, L. Mahendar, B. V. Ramulu and G. Satyanarayana, Synlett, 2012, 23, 375 CrossRef CAS PubMed; (c) A. G. K. Reddy and G. Satyanarayana, Tetrahedron, 2012, 68, 8003 CrossRef CAS PubMed; (d) A. G. K. Reddy, J. Krishna and G. Satyanarayana, Tetrahedron Lett., 2012, 53, 5635 CrossRef PubMed; (e) L. Mahendar, J. Krishna, A. G. K. Reddy, B. V. Ramulu and G. Satyanarayana, Org. Lett., 2012, 14, 628 CrossRef CAS PubMed; (f) J. Krishna, A. G. K. Reddy and G. Satyanarayana, Synlett, 2013, 24, 967 CrossRef CAS PubMed; (g) A. G. K. Reddy, J. Krishna and G. Satyanarayana, Tetrahedron, 2013, 69, 10098 CrossRef CAS PubMed; (h) J. Krishna, A. G. K. Reddy and G. Satyanarayana, Tetrahedron Lett., 2014, 55, 861 CrossRef CAS PubMed.
- (a) B. Suchand, J. Krishna, B. V. Ramulu, D. Dibyendu, A. G. K. Reddy, L. Mahendar and G. Satyanarayana, Tetrahedron Lett., 2012, 53, 3861 CrossRef CAS PubMed; (b) B. V. Ramulu, L. Mahendar, J. Krishna, A. G. K. Reddy, B. Suchand and G. Satyanarayana, Tetrahedron, 2013, 69, 8305 CrossRef CAS PubMed.
- B. V. Ramulu, A. G. K. Reddy and G. Satyanarayana, Synlett, 2013, 24, 868 CrossRef CAS PubMed.
-
 .
.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/C4RA00048J/ |
| This journal is © The Royal Society of Chemistry 2014 |