Enhanced electrochemical performances of FeOx–graphene nanocomposites as anode materials for alkaline nickel–iron batteries
Wei Jiangad,
Fei Liangab,
Jianwei Wangad,
Lei Suac,
Yaoming Wu*ab and
Limin Wang*ab
aState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, CAS, Changchun 130022, China. E-mail: lmwang@ciac.ac.cn; ymwu@ciac.ac.cn; Fax: +86-0431-85262836; Tel: +86-0431-85262447/04
bChangzhou Institute of Energy Storage Materials and Devices, Changzhou 213000, China
cFaculty of Chemistry, Northeast Normal University, Changchun 130024, China
dUniversity of Chinese Academy of Science, Beijing 130049, China
First published on 7th March 2014
Abstract
A new type of graphene-based FeOx nanocomposites have been synthesized by high temperature solid-state reaction using FeC2O4·2H2O. The synthesis conditions are optimized by thermogravimetric analysis of the precursor. When evaluated as anode material for the alkaline nickel–iron battery, the FeOx–graphene nanocomposites deliver a high specific capacity of 552.1 mA h g−1 at a current density of 200 mA g−1 and retain 91% of the initial capacity after 100 cycles. Furthermore, the hybridized FeOx–graphene materials undergo only 26% capacity decay when the discharge current density is changed from 200 mA g−1 to 1000 mA g−1. The enhanced cycling and high discharge rate performance derives from the high specific surface area of iron oxide nanoparticles and particular electric conductivity of graphene. This study suggests a safe, inexpensive and powerful rechargeable iron electrode, enabling the promising prospect of large-scale energy storage based on the aqueous iron-based rechargeable battery.
1. Introduction
Rechargeable batteries are especially suitable for large-scale storage of electrical energy because of their high energy efficiency and scalability.1–3 The lithium ion battery, lead-acid battery and nickel–metal hydride (Ni–MH) battery have played major roles in various application fields. The relatively low cost, eco-friendliness, as well as enhanced safety requirements have also attracted increasing attention to rechargeable batteries. The safety of the lithium ion battery, with its organic electrolytes and the high activity of the Li contained in the electrode materials,4–6 has been questioned by most researchers. In addition, the toxicity of the lead-acid battery and the high cost limitation of the Ni–MH battery are seemingly insurmountable problems. Thus, there is an urgent need for promising alternatives that could be constructed using inexpensive materials in relatively safe aqueous electrolytes.Developed around the 1900s by Waldemar Jungner and Thomas Edison, the nickel–iron battery used to be widely discussed for large-scale energy storage.7–9 With Ni(OH)2 as the cathode and iron as the anode, the century-old Ni–Fe battery was long considered to be one of the most promising secondary batteries.9 Presently, Ni–Fe energy storage systems have been widely employed in the electricity grid owing to their low cost, durability, eco-friendliness and safety. Nevertheless, the high self-discharge,10 relatively low energy efficiency,11,12 and especially the low power density13 of the iron anode in aqueous alkaline media are the principal drawbacks seriously restricting further utilization of the iron battery. Because of the passivation14,15 of the iron electrode surface, a practical capacity of only about one-third of the theoretical value (962 mA h g−1) can be achieved. The formation of a passive layer also leads to poor performance at high discharge rates. It is worthwhile to significantly improve the electrochemical performances of the iron electrode at high discharge rates, aimed at applications in hybrid electric vehicles (HEV) and electric vehicles (EV) where high current response is needed.
Graphene-based composites have been intensively explored in a wide range of applications,16,17 including supercapacitors,18 fuel cells,19 photovoltaic devices,20 photocatalysis,21 and batteries.22–26 In a previous study, Wang27 reported a strongly coupled FeOx–graphene hybrid as an anode for an ultrafast nickel–iron battery; this successfully increased the charging and discharging rates by nearly 1000-fold over traditional Ni–Fe batteries. In the present study, a novel hybridized FeOx–graphene anode material was prepared through a relatively simple high temperature solid-state reaction process instead of by hydrothermal synthesis. The iron oxide particles recrystallized on the reduced graphene sheets during the high temperature decomposition process. Direct growth of iron nanoparticles on graphene sheets provided a good contact between the FeOx nanoparticles and the two-dimensional network of graphene, thereby realizing efficient conduction of charge carriers and enhancing the structural stability. Consequently, FeOx–graphene composites can afford excellent electrochemical performances at a high discharge current density.
2. Experimental section
2.1 Synthesis of hybridized FeOx–graphene materials
Graphene oxide (GO) was made by a modified Hummers method28 using graphite powder (200 mesh, 99.9999%, Sigma-Aldrich). FeOx–graphene hybrids were synthesized by a ball milling method followed by a high temperature solid-state reaction process. Ferrous oxalate dihydrate (99.99%, Sigma-Aldrich) (FOD) and the as-prepared GO (10 mg mL−1) were mixed based on the mass ratio (mFOD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mGO = 100:1.0, 100:1.5 and 100:2.0), and 5% glucose was added as the reducing agent; ethanol (5 mL) was used as the dispersing agent. The raw materials were then sequentially ball-milled under an Ar atmosphere using a spex800 ball mill machine for 6 h with the ball-to-powder weight of 10:1. The intermediate products were kept at 80 °C for 12 h for intensive drying under vacuum. After that, the obtained precursors were heated at 350 °C for 0.5 h, then at 400 °C for 6 h, and finally, calcined at 700 °C for 16 h under an Ar atmosphere to produce the final FeOx–graphene composites. For comparison, FeOx composites were also prepared by a similar procedure in the absence of GO.
:mGO = 100:1.0, 100:1.5 and 100:2.0), and 5% glucose was added as the reducing agent; ethanol (5 mL) was used as the dispersing agent. The raw materials were then sequentially ball-milled under an Ar atmosphere using a spex800 ball mill machine for 6 h with the ball-to-powder weight of 10:1. The intermediate products were kept at 80 °C for 12 h for intensive drying under vacuum. After that, the obtained precursors were heated at 350 °C for 0.5 h, then at 400 °C for 6 h, and finally, calcined at 700 °C for 16 h under an Ar atmosphere to produce the final FeOx–graphene composites. For comparison, FeOx composites were also prepared by a similar procedure in the absence of GO.
2.2 Characterization
Thermogravimetric analysis of the FeOx–graphene precursor was investigated using a TG/DSC (SDT-2960, USA) apparatus with a temperature range 100–700 °C (10 °C min−1) under an Ar atmosphere. Further, the composition and phase purity of the as-prepared samples were characterized by X-ray diffraction (XRD) (D8 Focus, Bruker, Germany). Scanning electron microscopy (SEM) was carried out on an S-4800 instrument (Hitachi, Japan). Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were carried out on a Hitachi-600 instrument at an acceleration voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) measurement was performed on a ESCALAB MK II XPS spectrometer to analyse the surface composition, using monochromated Al Kα X-rays. Cyclic voltammetry (CV) measurements were performed using a VMP3 Electrochemical Workstation (Bio-logic Inc.) in the range of −0.4 to −1.4 V on a three-electrode configuration, where mercury/mercuric oxide was used as the reference electrode (E0 = +0.098 V vs. the normal hydrogen electrode) and Ni(OH)2 was used as the counter electrode. The electrolyte was 8 M KOH and 1 M LiOH mixed aqueous solution.2.3 Preparation of working electrodes and electrochemical measurements
The anode electrode typically contained 92 wt% of the synthesized FeOx–graphene materials, 5 wt% of bismuth oxide, and 3 wt% of hydroxypropyl methyl cellulose (HPMC). After the mixture was dispersed in D.I. water, the pastes were loaded onto a circular Ni foam current collectors (r = 8 mm, 110 ppi). The substrates were dried at 80 °C for 6 h in vacuum and then compressed to 0.5 mm thickness before measurement. Subsequently, the working electrodes were tested in a miniature cell, using two sintered nickel electrodes as cathode to achieve excess capacity and non-woven polypropylene cloth as separator. In accordance with the relevant literature,29 we chose 8 M KOH aqueous solution including 1 M LiOH as the electrolyte. Finally, electrochemical measurements were carried out using a LAND battery-test instrument (Wuhan, China) at room temperature. The specific capacity and cycle life at different current densities were tested to evaluate the performance of the hybridized materials.3. Results and discussion
TG-DSC analysis (Fig. 1) of the ball-milled precursor was carried out to investigate the solid-phase reaction process of the FeOx–graphene precursor within the temperature range 100 °C to 700 °C at a rate of 10 °C min−1. The results indicate three significant weight loss regions. The first region (150–200 °C) is related to the dehydration of the hydrates. The second plateau of weak weight loss observed at approximately 330 °C is attributed to the GO reduction process, on the basis of weight loss ratio. Subsequently, the thermal decomposition of FeC2O4 causes the endothermic peak at nearly 400 °C (ref. 30) followed by the glucose carbonization at above 500 °C.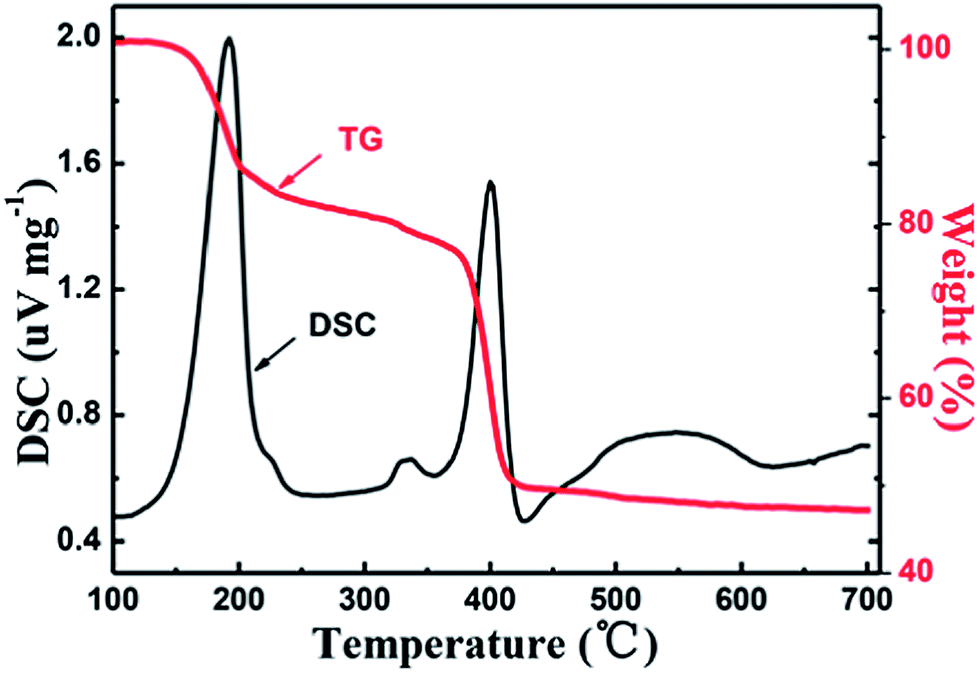 | ||
| Fig. 1 TG-DSC curve of the FeOx–graphene (mFOD:mGO = 100:1.5) precursor. | ||
Fig. 2 shows the XRD patterns of the FeOx composites prepared using different weight ratios of FOD and GO. The narrow sharp peaks indicate good crystallization of the samples. It is noteworthy that the FeOx grown on the reduced GO sheet was a mixture of Fe3O4 and FeO. For the sample prepared using mFOD:mGO = 100:2 in particular, it is obvious that no FeO peaks are detected in the product, indicating that the content of FeO particles decreased with the increase of GO. It can be assumed that the FeO particles were further oxidized by the resultant water and by the oxidizing agent produced along with the reduction process of GO. In brief, the decomposition process of the FeC2O4·2H2O can be expressed as
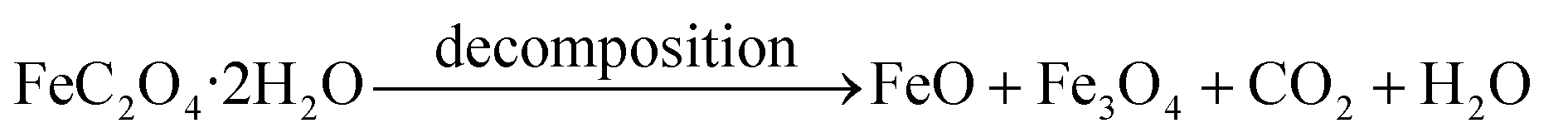 | (1) |
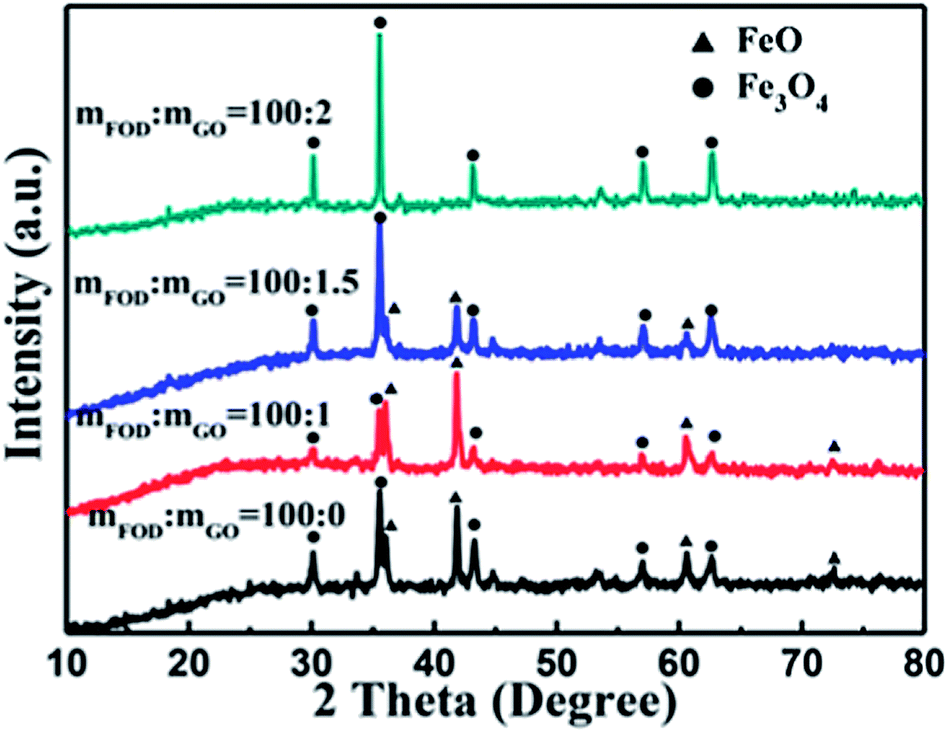 | ||
| Fig. 2 XRD patterns of FeOx–graphene composites prepared using different weight ratios of FOD and GO. | ||
To further investigate the chemical structure of the iron particles, typical XPS analysis was performed; the results are shown in Fig. 3. In the XPS Fe 2p spectrum (Fig. 3a), two peaks at 711.1 eV and 724.5 eV are detected, which are assigned to Fe 2p3/2 and Fe 2p1/2 binding energies. The Fe 2p3/2 electron-binding energy observed in Fig. 3a reveals that the compound FeOx contains mostly Fe3O4. FeO was barely detected in the XPS spectrum, possibly owing to its trace content and graphene covering. The deconvoluted O 1s XPS spectrum (Fig. 3b) shows two peaks at 530.1 eV and 531.9 eV, indicating the presence of O2− and OH−, respectively. These results indicate that the iron particles exist in a mixed state of iron oxides and a small amount of intermediate product FeOOH produced during the FeC2O4·2H2O pyrolysis. The characteristic peak of graphene at 284.8 eV is observed in the XPS C 1s spectrum (Fig. 3c), which confirmed the existence of reduced graphene oxide (rGO). The high-temperature reaction processes of FeOx recrystallization and graphene oxide reduction contribute to the intensive mixing between iron oxides and graphene.
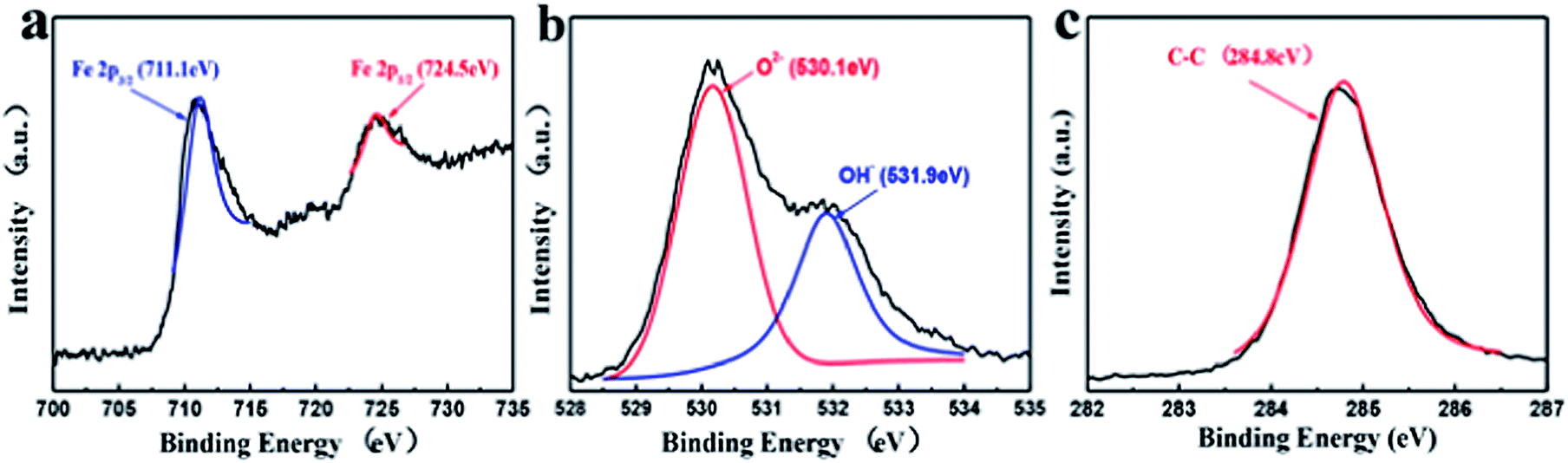 | ||
| Fig. 3 Deconvoluted Fe 2p (a), O 1s (b) and C 1s (c) XPS spectra of FeOx–graphene hybrids (mFOD:mGO = 100:1.5). | ||
Raman spectroscopy is widely used to evaluate the reduction degree of GO. The Raman spectra of graphene oxide and FeOx–graphene composites are plotted in Fig. 4. Characteristic peaks of FeOx–graphene materials at around 1340 cm−1 (D band) and around 1590 cm−1 (G band) are detected. The intensity ratio of D and G peaks (ID/IG) reveals the disorder density of carbon materials. As shown in Fig. 4, compared with GO (ID/IG = 0.86), the ID/IG of FeOx–graphene is increased to nearly 1.10 as a result of removal of the oxygen-containing functional groups during the GO reduction progress. The analysis of Raman spectra indicates that GO in the composite is reduced sufficiently.
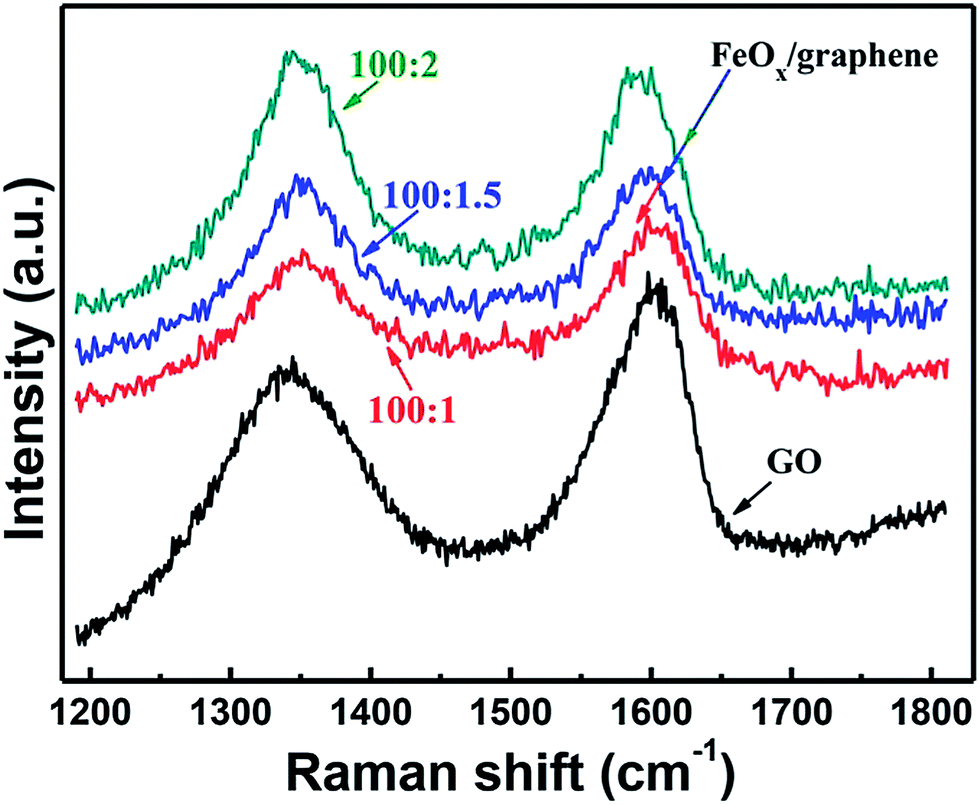 | ||
| Fig. 4 Raman spectra of graphene oxide and FeOx–graphene composites prepared using different weight ratios of FOD and GO. | ||
The SEM image of the FeOx–graphene composite with mFOD:mGO = 100:1.5 (Fig. 5a) reveals that the FeOx nanoparticles coat evenly on the graphene sheet. Most of the nanoparticles are spherical with diameters ranging from 20 to 100 nm, compared with the blank sample (Fig. 5b), which are in the size range of 1–3 μm, and are strongly aggregated. The direct growth of inorganic nanomaterials on mildly oxidized nanocarbon materials affords strong covalent coupling between inorganic nanocrystals and carbon materials27 that impedes the nanoparticles from crystallization. In Fig. 5c, the TEM image clearly shows the nanoparticles grown on the graphene (GE) sheets. The HRTEM image of the FeOx–graphene composites (Fig. 5d) reveals the lattice spacing of 0.47 nm corresponding to the (111) plane of Fe3O4, which proves the existence of Fe3O4 in the FeOx composites.
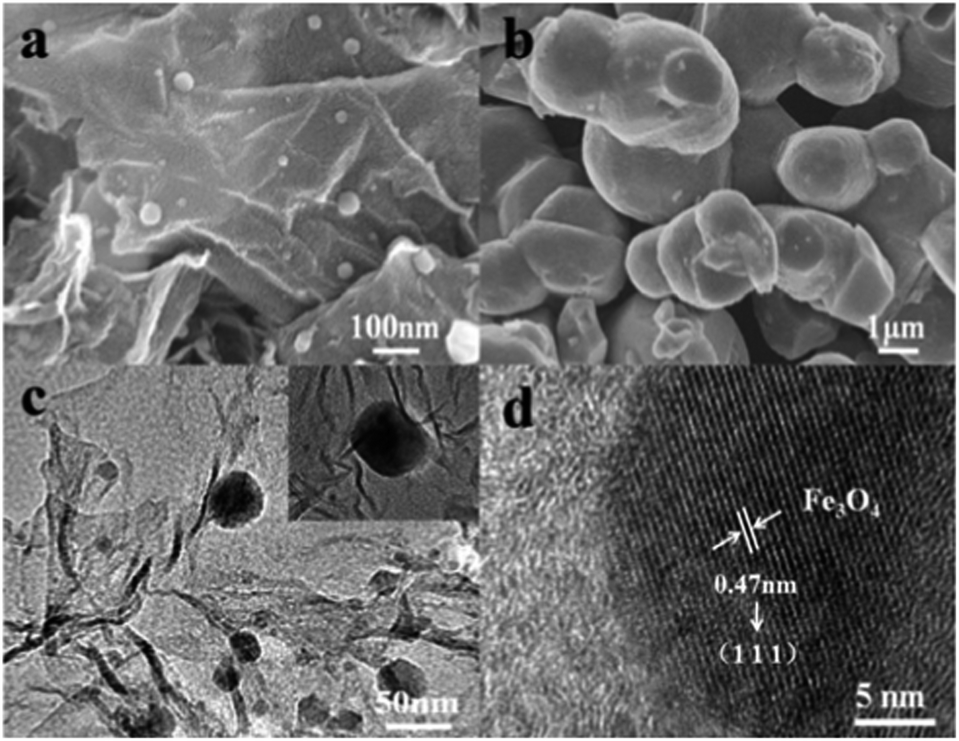 | ||
| Fig. 5 SEM images of the FeOx–graphene composites, (a) mFOD:mGO = 100:1.5 (b) mFOD:mGO = 100:0; (c) TEM and (d) HRTEM images of the FeOx–graphene (mFOD:mGO = 100:1.5). | ||
Cyclic voltammograms (CVs) were obtained to investigate the oxidation–reduction behaviour of the FeOx–graphene system. Fig. 6a shows the CV curves of the different FeOx electrodes. The FeOx–graphene sample with mFOD:mGO = 100:1.5 exhibits a relatively better reversible electrochemical oxidation–reduction process. Even at higher scan rates (Fig. 6b), the strong oxidation and reduction peaks are still observed, implying the excellent reversibility of the hybrid materials at high current density. According to previous studies,31,32 multiple peaks should be observed in the CV curves as a result of the redox reaction of the iron electrode. But, in particular, the CV curve only displays one overlapping redox peak at −1.08 V, mainly involved with the conversion of Fe2+ to Fe. Two obvious oxidation peaks with higher potential and stronger intensity appear at −0.87 V and −0.55 V , representing Fe/Fe2+ and Fe2+/Fe3+, respectively, as shown by the following reactions:32
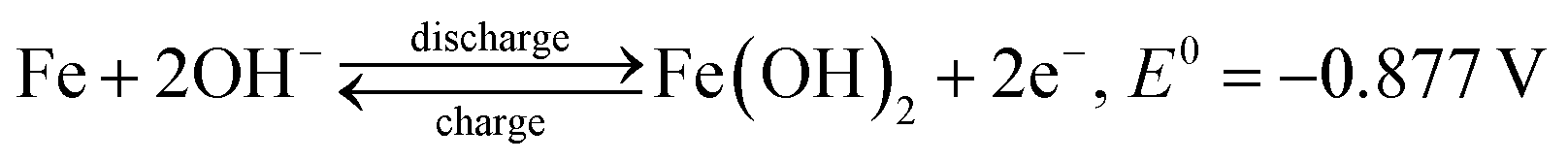 | (2) |
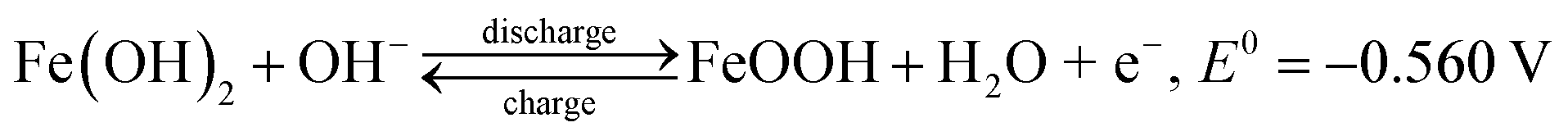 | (3) |
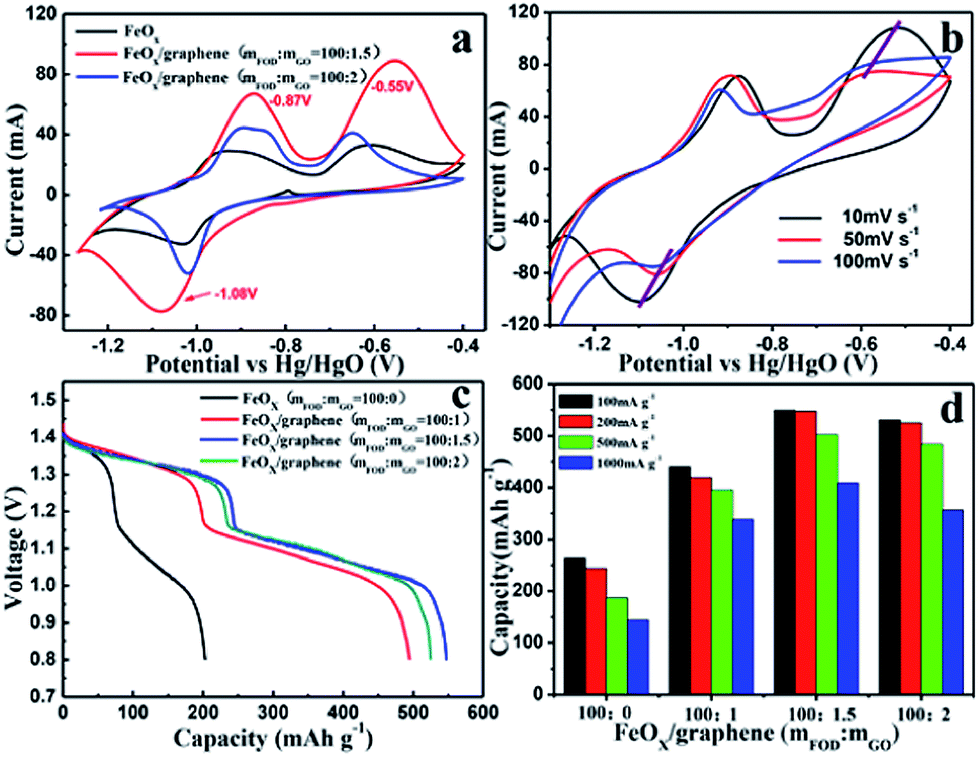 | ||
| Fig. 6 (a) CV curves of FeOx–graphene hybrids (mFOD:mGO = 100:0, 100:1.5, 100:2) at a scan rates of 5 mV s−1; (b) CV curves of FeOx–graphene hybrids (mFOD:mGO = 100:1.5) at 10 mV s−1, 50 mV s−1, and 100 mV s−1. (c) Typical discharge curves of the FeOx–graphene hybrids (mFOD:mGO = 100:1.5) at a constant current density of 200 mA g−1. (d) Discharge capacity of the four samples at different discharge rate (100 mA g−1, 200 mA g−1, 500 mA g−1 and 1000 mA g−1). | ||
In order to investigate the specific capacity of the FeOx–graphene composites, charge–discharge cycles at a discharge current density of 200 mA g−1 were carried out. Two discharge plateaus are observed in the discharge process (Fig. 6c), which can be attributed to the reactions Fe → Fe2+ and Fe2+ → Fe3+. Obviously, the discharge capacities of FeOx–graphene composites are more than twice that of the blank sample. In particular for the samples with mFOD:mGO = 100:1.5, the maximum discharge capacity can be maintained at 552.1 mA h g−1 (compared with 222.6 mA h g−1 of the FeOx material without graphene). The electrochemical performance of the FeOx–graphene hybrids was further evaluated by high-rate discharge measurement. Fig. 6d shows the discharge capacity of the FeOx–graphene hybrids at different current densities from 100 mA g−1 to 1000 mA g−1. As expected, our FeOx–graphene composites display excellent high-rate performance. Particularly for the optimal sample (mFOD:mGO = 100:1.5), the specific capacity changed from 549.4 mA h g−1 to 408.5 mA h g−1, based on the discharge current densities of 100 mA g−1 and 1000 mA g−1, respectively. The outstanding electrochemical performance can be attributed to the following reasons: first, the size of the FeOx particles decreased to a large extent because of the strong coupling with graphene during decomposition–crystallization progress, which contributes to a larger efficient surface area; and second, the GE layers can enhance the electron transfer through the nano-sized active materials during charging and discharging.
Fig. 7a shows the cycle stability of the FeOx–graphene composite (mFOD:mGO = 100:1.5) electrode at discharge current densities of 200 mA g−1 to 1000 mA g−1. About 80% of the initial capacity is maintained after 100 charge–discharge cycles at a current density of 200 mA g−1. Additionally, even at a high discharge current density of 1000 mA g−1, the capacity decay remains as low as 10% after 100 cycles. It is apparent that the particular electronic conductivity, the salutary space effect, as well as the mechanical property of GE leads to the outstanding cycle performance of the FeOx–graphene electrode.27,33,34 We obtained the XRD pattern of a sample in the discharged state (Fig. 7b) and SEM images of the two samples in the charged state (Fig. 7c and d) to further investigate the morphology and microstructure variation of the composites after 100 charge–discharge cycles. Fig. 7b clearly shows the existence of Fe3O4, which is produced during the discharge reaction. As revealed in SEM images, the reversible capacity decay results from the growth of iron oxide particles during cycles. Additionally, the graphene-based FeOx particles grow evenly compared with the serious aggregation of the blank FeOx particles during the dissolution–deposition process. In other words, FeOx–graphene composites have high structural stability leading to excellent cycle performance.
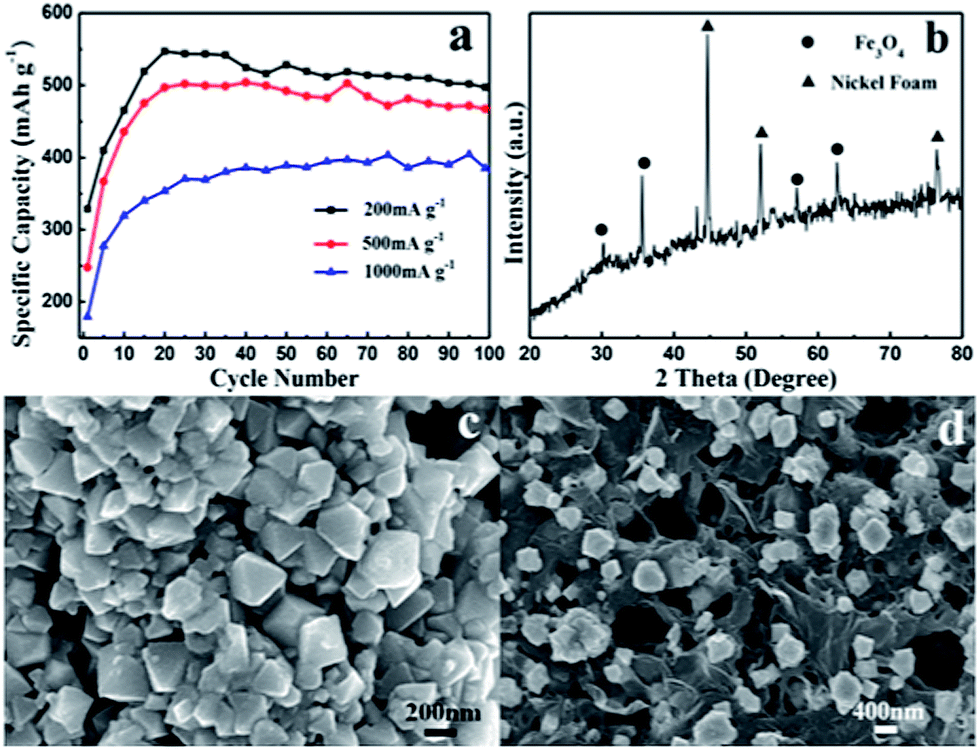 | ||
| Fig. 7 (a) Cycling performance of FeOx–graphene hybrids (mFOD:mGO = 100:1.5) at different discharge current densities (200 mA g−1, 500 mA g−1 and 1000 mA g−1); (b) XRD patterns of FeOx–graphene hybrids (mFOD:mGO = 100:1.5) in the discharged state after 100 cycles; SEM images of (c) blank sample and (d) FeOx–graphene (mFOD:mGO = 100:1.5) sample in the charged state after 100 cycles. | ||
4. Conclusions
In summary, a new type of FeOx nanocomposites grown on graphene sheets were successfully synthesized via a simple high-temperature solid-state reaction. The results indicated that the FeOx particles were composed of Fe3O4 and FeO with particle size around 100 nm. The FeOx–graphene nanocomposites delivered an initial discharge capacity of 552.1 mA h g−1 at a current density of 200 mA g−1. Owing to the electrochemical activity and mechanical properties that result from strong coupling between iron-oxide nanoparticles and the graphene layer, the FeOx–graphene composites exhibited enhanced high discharge rate capability and cycling stability. The research presented here demonstrates a simple and promising method of synthesizing iron anode materials for improving the power density of rechargeable iron battery for large-scale energy storage, as well as for electric vehicles and hybrid electric vehicles.Acknowledgements
This study was supported by the National Nature Science Foundation of China (Grant nos 20111061 and 21373198).Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, J. Power Sources, 2011, 196, 6688–6694 CrossRef CAS.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- G. Halpert, J. Power Sources, 1984, 12, 177–192 CrossRef CAS.
- A. K. Shukla, M. K. Ravikumar and T. S. Balasubramanian, J. Power Sources, 1994, 51, 29–36 CrossRef CAS.
- C. Chakkaravarthy, P. Periasamy, S. Jegannathan and K. I. Vasu, J. Power Sources, 1991, 35, 21–35 CrossRef CAS.
- C. A. C. Souza, I. A. Carlos, M. Lopes, G. A. Finazzi and M. R. H. de Almeida, J. Power Sources, 2004, 132, 288–290 CrossRef CAS.
- A. K. Manohar, S. Malkhandi, B. Yang, C. G. Yang, G. K. S. Prakash and S. R. Narayanan, J. Electrochem. Soc., 2012, 159, A1209–A1214 CrossRef CAS.
- S. Malkhandi, B. Yang, A. K. Manohar, G. K. S. Prakash and S. R. Narayanan, J. Am. Chem. Soc., 2012, 135, 347–353 CrossRef PubMed.
- P. Periasamy, B. R. Babu and S. V. Iyer, J. Power Sources, 1996, 58, 35–40 CrossRef CAS.
- W. C. He, H. B. Shao, Q. Q. Chen, J. M. Wang and J. Q. Mang, Acta Phys.-Chim. Sin., 2007, 23, 1525–1530 CrossRef CAS.
- A. K. Manohar, C. G. Yang, S. Malkhandi, B. Yang, G. K. S. Prakash and S. R. Narayanan, J. Electrochem. Soc., 2012, 159, A2148–A2155 CrossRef CAS.
- X. Huang, X. Y. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- J. H. Zhu, M. J. Chen, Q. L. He, L. Shao, S. Y. Wei and Z. H. Guo, RSC Adv., 2013, 3, 22790–22824 RSC.
- W. H. Shi, J. X. Zhu, D. H. Sim, Y. Y. Tay, Z. Y. Lu, X. J. Zhang, H. Zhang, H. H. Hng and Q. Yan, J. Mater. Chem., 2011, 21, 3422–3427 RSC.
- Y. Li, L. Tang and J. Li, Electrochem. Commun., 2009, 11, 846–849 CrossRef CAS.
- Z. Yin, S. Wu, X. Zhou, X. Huang, Q. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 307–312 CrossRef CAS PubMed.
- H. Liu, S. Ryu, Z. Chen, M. L. Steigerwald, C. Nuckolls and L. E. Brus, J. Am. Chem. Soc., 2009, 131, 17099–17101 CrossRef CAS PubMed.
- J. Zhu, T. Zhu, X. Zhou, Y. Zhang, X. W. Lou, X. Chen, H. Chen, H. Zhang, H. H. Hng, J. Ma and Q. Yan, Nanoscale, 2011, 3, 1084–1089 RSC.
- Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- S. Yang, X. Feng, S. Ivanovici and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 8408–8411 CrossRef CAS PubMed.
- H. Wang, L.-F. Cui, Y. Yang, H. S. Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS PubMed.
- Q. H. Wang, L. F. Jiao, H. M. Du, Y. J. Wang and H. T. Yuan, J. Power Sources, 2014, 245, 101–106 CrossRef CAS.
- H. L. Wang, Y. Y. Liang, M. Gong, Y. G. Li, W. Chang, T. Mefford, J. G. Zhou, J. Wang, T. Regier, F. Wei and H. J. Dai, Nat. Commun., 2012, 3, 917–924 CrossRef PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Liu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- C. Y. Kao and K. S. Chou, J. Power Sources, 2010, 195, 2399–2404 CrossRef CAS.
- Z. D. Fang and D. J. Wang, Chin. J. Inorg. Chem., 2005, 21, 1682–1686 CAS.
- P. Periasamy, B. R. Babu and S. V. Iyer, J. Power Sources, 1996, 58, 35–40 CrossRef CAS.
- K. Vijayamohanan, T. S. Balasubramanian and A. K. Shukla, J. Power Sources, 1991, 34, 269–285 CrossRef CAS.
- A. M. Rafiee, J. Rafiee, Z. Wang, H. H. Song, Z. Z. Yu and N. Koratkar, ACS Nano, 2009, 3, 3884–3890 CrossRef PubMed.
- S. L. Chou, J. Z. Wang, M. Choucair, H. K. Liu, J. A. Stride and S. X. Dou, Electrochem. Commun., 2010, 12, 303–306 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |